Государственное санитарно-эпидемиологическое нормирование
Российской Федерации
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
ОПРЕДЕЛЕНИЕ
ОСТАТОЧНЫХ
КОЛИЧЕСТВ ПЕСТИЦИДОВ
В ПИЩЕВЫХ ПРОДУКТАХ,
СЕЛЬСКОХОЗЯЙСТВЕННОМ СЫРЬЕ
И ОБЪЕКТАХ ОКРУЖАЮЩЕЙ СРЕДЫ
Сборник
методических указаний
МУК
4.1.2062 - 4.1.2074-06
Москва, 2009
1. Сборник подготовлен Федеральным научным центром гигиены им. Ф.Ф. Эрисмана (академик РАМН, проф. В.Н. Ракитский, проф. Т.В. Юдина); при участии специалистов Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека. Разработчики методов указаны в каждом из них.
2. Рекомендованы к утверждению Комиссией по государственному санитарно-эпидемическому нормированию при Федеральной службе по надзору в сфере защиты прав потребителей и благополучия человека.
3. Утверждены Главным государственным санитарным врачом Российской Федерации, Первым заместителем Министра здравоохранения Российской Федерации, академиком РАМН Г.Г. Онищенко.
4. Введены впервые.
Содержание
|
УТВЕРЖДАЮ |
|
Руководитель Федеральной службы |
|
по надзору в сфере защиты |
|
прав потребителей и благополучия |
|
человека, Главный государственный |
|
санитарный врач Российской Федерации |
|
Г.Г. Онищенко |
|
5 мая 2006 г. |
|
МУК 4.1.2063-06 |
|
Дата введения: с 1 июля 2006 г. |
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
ПО ОПРЕДЕЛЕНИЮ ОСТАТОЧНЫХ КОЛИЧЕСТВ ТРИАСУЛЬФУРОНА
В ЗЕРНЕ ХЛЕБНЫХ ЗЛАКОВ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ
ЖИДКОСТНОЙ ХРОМАТОГРАФИИ
1. Вводная часть
Торговое наименование: Дукат.
Фирма регистрант: ООО НПП «Агрусхим».
Действующее вещество: триасульфурон.
Структурная формула:
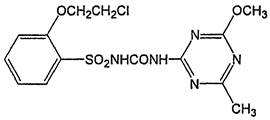
1-[2-(2-хлорэтокси)фенилсульфонил]-3-(6-метил-4-метокси-1,3,5-триазин-2-ил)мочевина (IUPAC).
2-(2-хлорэтокси)-[[(6-метил-4-метокси-1,3,5-триазин-2-ил)амино]карбонил]бензолсульфонамид (СА).
Мол. масса: 401,8.
Брутто формула: C14H16ClN5O5S.
Химически чистый триасульфурон представляет собой белый порошок с температурой плавления 178,1 °С, давлением паров < 2×10-3 мРа (25 °С).
Коэффициент распределения в системе н-октанол-вода KowlgP = 1,1 (pH 5), -0,59 (pH 7), -1,8 (pH 9) (25 °С).
Растворимость (г/дм3): в воде 0,032 (pH 5), 0,815 (pH 7), 13,5 (pH 8,4) при 25 °С; дихлорметане - 36, ацетоне - 14, этилацетате - 4,3, этаноле - 0,42, толуоле - 0,3, н-октаноле - 0,13, гексане - 0,00004 (25 °С).
Стабилен на воздухе до 140 °С, в нейтральных и щелочных растворах при 25 °С.
Группа токсичности по ЕПА - IV, ВОЗ - U; Острая пероральная токсичность LD50 для крыс и мышей > 5000 мг/кг. Дермальная токсичность для крыс > 2000 мг/кг.
Область применения: селективный гербицид, адсорбируется листьями, корнями и быстро попадает в меристему. Используется для контроля широколиственных сорняков на зерновых. Гигиенические нормативы для триасульфурона в России: МДУ в зерне хлебных злаков - 0,1 мг/кг.
2. Методика определения триасульфурона в зерне хлебных злаков методом ВЭЖХ
2.1. Основные положения
2.1.1. Область применения и принцип метода
Настоящий документ устанавливает методику определения остаточных количеств триасульфурона в пробах зерна хлебных злаков в диапазоне концентраций 0,01 - 0,1 мг/кг.
Методика основана на определении триасульфурона методом ВЭЖХ с использованием УФ детектора после его извлечения из образцов водно-ацетоновой смесью с последующей очисткой перераспределением между двумя несмешивающимися растворителями и на колонке с силикагелем.
2.1.2. Метрологическая характеристика метода
Диапазон определяемых концентраций 0,01 - 0,1 мг/кг.
Нижний предел определения триасульфурона в зерне 0,01 мг/кг.
Полнота определения 80 % (коэффициент извлечения K = 0,8),
Границы суммарной относительной погрешности результата измерений ∆ ± 12 % при доверительной вероятности Р = 0,95 (n = 20).
Полнота определения триасульфурона в зерне зерновых колосовых культур (N = 5 для каждой концентрации):
|
Среда |
Внесено, мг/кг |
Найдено, мг/кг |
Стандартное отклонение, ± |
Полнота определения, % |
|
1 |
2 |
3 |
4 |
5 |
|
Зерно |
0,01 |
0,0079 |
0,00069 |
79,0 |
|
|
0,025 |
0,0202 |
0,0017 |
80,7 |
|
|
0,05 |
0,0396 |
0,0033 |
79,2 |
|
|
0,10 |
0,0828 |
0,0075 |
82,8 |
|
Среднее |
|
|
|
80,4 |
2.1.3. Избирательность метода
Присутствие других пестицидов, близких по химическому строению и области применения определению не мешает.
2.2. Реактивы и материалы
Ацетон, осч, ТУ 6-09-3513-86.
Ацетонитрил для ВЭЖХ, «В-230НМ» или х.ч., ТУ 6-09-3534-87.
Бумажные фильтры «красная лента», ТУ 6.091678-86.
Вода бидистиллированная, деионизированная, ГОСТ 6709-79.
Калий углекислый, х.ч., ГОСТ 4221-76.
Калий фосфорнокислый 2-замещенный, 3-водный, чда, ГОСТ 2493-75.
Калия перманганат, ГОСТ 20490-75.
Кальция хлорид, х.ч., ГОСТ 4161-77.
Кислота ортофосфорная, имп. (Ferak, Германия) или хч, ГОСТ 6552-80; 2М и 0,005М водные растворы.
Кислота серная, х.ч., ГОСТ 4204-77.
Трибенурон-метил, аналитический стандарт с содержанием д.в. 98,2 % (Sigma-Aldrich).
Натрий двууглекислый, ГОСТ 83-79.
Натрий сернокислый безводный, ч., ГОСТ 4166-76, свежепрокаленный.
Натрия гидроксид, хч., ГОСТ 4328-77.
н-Гексан, х.ч., ТУ 2631-003-05807999-98, свежеперегнанный.
Подвижная фаза для ВЭЖХ: смесь ацетонитрил - 0,01М Н3РО4 (35:65, по объему).
Силикагель для колоночной хроматографии 60 (0,040 - 0,063 mm) (Merck, Германия).
Стекловата.
Фосфора пентоксид, ч., МРТУ 6-09-5759-69.
Элюент № 1 для колоночной хроматографии: смесь гексан - этилацетат (50:50, по объему).
Элюент № 2 для колоночной хроматографии: смесь гексан - этилацетат (10:90, по объему).
Этиловый эфир уксусной кислоты, ч.д.а., ГОСТ 22300-76.
2.3. Приборы и посуда
Жидкостный хроматограф LC-10A фирмы Shimadzu с УФ детектором (SPD-10A) или другой с аналогичными характеристиками.
Аналитическая колонка Supelcosil С-18 (150×4,6) мм, 5 мкм (Sigma-Aldrich).
Предколонка Supelcosil С-18.
Весы аналитические ВЛА-200, ГОСТ 24104-2001 или аналогичные.
Установка ультразвуковая «Серьга», ТУ 3.836.008.
Мельница лабораторная зерновая ЛМЗ, ТУ 1-01-0593-79.
Ротационный испаритель вакуумный ИР-1М, ТУ 25-11-917-74 или аналогичный.
Бидистиллятор.
РН-метр универсальный ЭВ-74, ГОСТ 22261-76.
Насос водоструйный, МРТУ 42861-64.
Колбы плоскодонные на шлифах КШ500 29/32 ТС, ГОСТ 10384-72.
Колбы круглодонные на шлифах КШ50 29/32 ТС, ГОСТ 10384-72.
Воронки лабораторные В-75-110, ГОСТ 25336-82.
Воронки делительные ВД-3-500, ГОСТ 8613-75.
Цилиндры мерные на 100, 250 и 1000 см3, ГОСТ 1774-74.
Колбы мерные на 25, 50, 100 и 1000 см3, ГОСТ 1770-74.
Пипетки на 1, 2, 5, 10 см3, ГОСТ 22292-74.
Колонки стеклянные (25×1) см.
Допускается применять другие вспомогательное оборудование, материалы и реактивы с техническими и метрологическими характеристиками не хуже указанных.
2.4. Отбор и хранение проб
Отбор проб производится в соответствии с ГОСТ Р 50436-92 (ИСО 950-79) «Зерновые. Отбор проб зерна». Пробы зерна для определения остатков в урожае хранят в бумажной или тканевой упаковке при комнатной температуре. Перед анализом зерно доводят до стандартной влажности и измельчают на лабораторной мельнице.
2.5. Подготовка к определению
2.5.1. Подготовка и очистка реактивов и растворителей
Перед началом работы рекомендуется проверить чистоту применяемых органических растворителей. Для этого 100 см3 растворителя упаривают в ротационном испарителе при температуре 40 °С до объема 1,0 см3 и хроматографируют. При обнаружении мешающих определению примесей очистку растворителей производят в соответствии с типовыми методиками. Гексан и хлористый метилен встряхивают с небольшими порциями концентрированной серной кислоты до прекращения окрашивания свежей порции кислоты, затем промывают водой, 2 %-ным раствором гидроксида натрия и снова водой, после чего его сушат над гидроксидом натрия и перегоняют. Ацетон перегоняют над перманганатом калия и поташом (на 1 дм3 ацетона 10 г KМnO4 и 2 г K2СО3). Ацетонитрил сушат над пентоксидом фосфора и перегоняют; отогнанный растворитель повторно перегоняют над углекислым калием. Этилацетат промывают равным объёмом 5 %-ного раствора двууглекислого натрия, сушат над хлористым кальцием и перегоняют.
2.5.2. Кондиционирование колонки
Перед началом анализа колонку (Supelcosil С-18) кондиционируют в потоке подвижной фазы (1 см3/мин) до стабилизации нулевой линии в течение 1 - 2 часов.
2.5.3. Приготовление растворов
Для приготовления 2М раствора ортофосфорной кислоты 200 г 98 % (или 225 г 87 %) кристаллической Н3РО4 помещают в мерную колбу объемом 1 дм3, растворяют в 600 см3 дистиллированной воды и доводят объем до метки дистиллированной водой.
Для приготовления 0,01М раствора ортофосфорной кислоты 5,0 см3 2М раствора Н3РО4 вносят в мерную колбу на 1 дм3 и доводят до метки деионизированным бидистиллятом.
Для приготовления 0,2М раствора K2НРО4 45,6 г кристаллического калия фосфорнокислого двузамещенного трехводного помещают в мерную колбу на 1 дм3, растворяют при перемешивании в 600 см3 дистиллированной воды и доводят объем раствора до метки.
Для получения 50 %-го водного ацетона в колбе емкостью 1 дм3 смешивают 500 см3 ацетона с 500 см3 дистиллированной воды, используя мерные цилиндры.
Для приготовления подвижной фазы смешивают ацетонитрил с 0,01М раствором ортофосфорной кислоты в соотношении 35:65 по объему, используя мерные цилиндры.
Для приготовления элюента № 1 в колбе на 1000 см3 смешивают 500 см3 н-гексана и 500 см3 этилацетата. Для приготовления элюента № 2 в колбе на 1000 см3 смешивают 100 см3 н-гексана и 900 см3 этилацетата.
Приготовление стандартного и градуировочных растворов:
Берут точную навеску триасульфурона (50 мг), переносят в мерную колбу на 100 см3, растворяют навеску в ацетонитриле и доводят до метки. (Стандартный раствор с концентрацией 0,5 мг/см3). Градуировочные растворы с концентрациями 0,2, 0,5, 1,0 и 2,0 мкг/см3 готовят методом последовательного разбавления по объему, используя раствор подвижной фазы - смесь ацетонитрил - 0,01М ортофосфорная кислота (35:65, по объему).
Стандартный раствор можно хранить в холодильнике при температуре 0 - 4 °С в течение 1 месяца, градуировочные растворы - в течение суток.
При изучении полноты определения триасульфурона используют ацетонитрильные растворы вещества. Растворы внесения с концентрациями 0,2 и 2,0 мкг/см3 готовят из стандартного раствора с концентрацией 0,5 мг/см3 методом последовательного разбавления по объему ацетонитрилом.
2.5.4. Построение градуировочного графика
Для построения градуировочного графика (площадь пика - концентрация триасульфурона в растворе) в хроматограф вводят по 20 мм3 градуировочных растворов (не менее 3-х параллельных измерений для каждой концентрации, не менее 4-х точек по диапазону измеряемых концентраций). Затем измеряют площади пиков и строят график зависимости среднего значения площади пика от концентрации триасульфурона в градуировочном растворе (мкг/см3).
2.5.5. Подготовка колонки с силикагелем для очистки экстракта
В нижнюю часть стеклянной колонки длиной 25 см и внутренним диаметром 1 см помещают тампон из стекловаты, закрывают кран и вносят суспензию 5 г силикагеля в 30 см3 смеси гексан - этилацетат (50:50, по объему). Дают растворителю стечь до верхнего края сорбента. Колонку последовательно промывают 50 см3 элюента № 2 и 50 см3 элюента № 1 со скоростью 1 - 2 капли в секунду, после чего она готова к работе.
2.5.6. Проверка хроматографического поведения триасульфурона на колонке с силикагелем
В круглодонную колбу емкостью 10 см3 отбирают 0,1 см3 стандартного раствора триасульфурона с концентрацией 10 мкг/см3. Отдувают растворитель током теплого воздуха (температура не выше 40 °С), остаток растворяют в 5 см3 элюента № 1 и наносят на колонку. Колбу обмывают еще 5 см3 элюента № 1 и также вносят на колонку. Промывают колонку 50 см3 элюента № 1, затем 100 см3 элюента № 2 со скоростью 1 - 2 капли в секунду. Отбирают фракции по 10 см3 каждая, упаривают, остаток растворяют в 1 см3 подвижной фазы для ВЭЖХ (п. 2.5.3) и анализируют на содержание триасульфурона по п. 2.6.3.
Фракции, содержащие триасульфурон, объединяют, упаривают досуха, остаток растворяют в 1 см3 подвижной фазы для ВЭЖХ и вновь анализируют по п. 2.6.3. Рассчитывают содержание триасульфурона в элюате, определяя полноту вымывания вещества из колонки и необходимый для этого объем элюента.
Примечание: параметры удерживания триасульфурона и сопутствующих коэкстрактивных веществ могут меняться при использовании новой партии сорбента и растворителей.
2.6. Проведение определения
2.6.1. Определение триасульфурона в зерне
Навеску размолотого на лабораторной мельнице зерна массой 20 г помещают в коническую колбу емкостью 250 см3 и экстрагируют триасульфурон 50 см3 смеси ацетон-вода (1:1, по объёму) на ультразвуковой установке в течение 15 мин. Суспензию фильтруют через бумажный фильтр «красная лента». Экстракцию повторяют дважды порциями по 50 см3, выдерживая в ультразвуковой ванне каждый раз в течение 15 мин. Объединенный экстракт упаривают на ротационном испарителе при температуре бани не выше 40 °С до полного удаления ацетона. Объём водного остатка доводят до 100 см3 дистиллированной водой, добавляют к нему 50 см3 0,2М раствора K2НРО4.
Внимание! Отделение водного слоя следует производить только после полного расслоения жидкостей в делительной воронке.
Объединённый водный экстракт промывают в делительной воронке объёмом 500 см3 дважды гексаном порциями по 50 см3 (верхний органический слой отбрасывают) и дважды этилацетатом порциями по 50 см3 (верхний органический слой отбрасывают), встряхивая каждый раз делительную воронку в течение 2 - 3 мин. Водную фазу подкисляют 2М ортофосфорной кислотой до pH 3 и триасульфурон экстрагируют этилацетатом трижды по 30 см3, встряхивая воронку каждый раз по 2 - 3 мин*. Нижний водный слой отбрасывают. Объединенную органическую фазу фильтруют через слой безводного сульфата натрия (2 г), осушитель промывают 10 - 15 см3 этилацетата. Полученный раствор выпаривают досуха на роторном испарителе при температуре не выше 40 °С. Дальнейшую очистку экстракта проводят по пункту 2.6.2**.
__________
* В случае образования сравнительно стойких эмульсий для сокращения времени расслоения можно добавить в делительную воронку: на стадии промывки экстрактов гексаном - небольшое количество (до 10 см3) этилового спирта.
** В случаях, когда очистка экстрактов контрольных проб (п. 2.6.1) дает удовлетворительные результаты дополнительную очистку на колонке с силикагелем можно исключить.
2.6.2. Очистка на колонке с силикагелем
Сухой остаток в колбе, полученный при упаривании очищенных по п.п. 2.6.1 экстрактов зерна, количественно переносят двумя порциями по 5 см3 смеси гексан - этилацетат (50:50, по объему) в кондиционированную хроматографическую колонку (п. 2.5.5). Промывают колонку 50 см3 элюента № 1, которые отбрасывают. Триасульфурон элюируют 80 см3 элюента № 2, собирая элюат в грушевидную колбу емкостью 250 см3. Раствор выпаривают досуха на вакуумном ротационном испарителе при температуре не выше 40 °С. Сухой остаток растворяют в 1 см3 подвижной фазы для ВЭЖХ и 20 мм3 раствора вводят в жидкостный хроматограф.
2.6.3. Условия хроматографирования
Жидкостный хроматограф LC-10A фирмы Shimadzu с УФ детектором (SPD-10A) или другой с аналогичными характеристиками.
Рабочая длина волны 223 нм.
Колонка Supelcosil С-18 (150×4,6) мм, 5 мкм (Sigma-Aldrich).
Предколонка Waters Symmetry С-18 для защиты аналитической колонки.
Температура колонки 30 ± 1 °С.
Подвижная фаза: ацетонитрил - 0,01М раствор Н3РО4 в соотношении 35:65 (по объему).
Скорость потока элюента: 1 см3/мин.
Время удерживания триасульфурона 8,3 ± 0,1 мин.
2.6.4. Обработка результатов анализа
Количественное определение проводят методом абсолютной калибровки, содержание триасульфурона в образце зерна (X, мг/кг) вычисляют по формуле:
![]()
где S1 - площадь пика триасульфурона в стандартном растворе, мм;
S2 - площадь пика триасульфурона в анализируемой пробе, мм;
V - объем пробы, подготовленной для хроматографического анализа, см3;
Р - навеска анализируемого образца, г;
С - концентрация стандартного раствора триасульфурона, мкг/см3;
K - коэффициент извлечения, определяемый в эксперименте по внесению стандартного раствора триасульфурона в контрольный образец зерна.
Содержание остаточных количеств триасульфурона в анализируемом образце вычисляют как среднее из 2-х параллельных определений.
Образцы, дающие пики большие, чем стандартный раствор триасульфурона 2 мкг/см3, разбавляют подвижной фазой для ВЭЖХ.
3. Оформление результатов измерений
Результаты измерений содержания триасульфурона в образцах зерна представляют в виде:
(X ± ∆) мг/кг,
где ∆ - показатель точности результатов анализа (границы, в которых погрешность любого из совокупности результатов анализа, полученных при реализации методики, находится с принятой вероятностью Р = 0,95).
4. Требования техники безопасности
При проведении работы необходимо соблюдать требования безопасности, установленные для работ с токсичными, едкими, легковоспламеняющимися веществами (ГОСТ 12.1.005-88).
При выполнении измерений с использованием жидкостного хроматографа и работе с электроустановками соблюдать правила электробезопасности в соответствии с ГОСТ 12.1.019-79 и инструкциями по эксплуатации приборов.
Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004-91.
5. Требования к квалификации оператора
Измерения в соответствии с настоящей методикой может выполнять специалист-химик, имеющий опыт работы методом высокоэффективной жидкостной хроматографии, ознакомленный с руководством по эксплуатации жидкостного хроматографа, освоивший данную методику и подтвердивший экспериментально соответствие получаемых результатов нормативам контроля погрешности измерений по п. 6.
6. Контроль погрешности измерений
Оперативный контроль погрешности и воспроизводимости результатов измерений осуществляется в соответствии с ГОСТ Р ИСО 5725-1 - 6-2002 «Точность (правильность и прецизионность) методов и результатов измерений».
7. Разработчики
Долженко В.И., Цибульская И.А., Юзихин О.С. (ВНИИ защиты растений, Санкт-Петербург).