Федеральная служба по надзору в сфере защиты прав
потребителей
и благополучия человека
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение остаточных
количеств химических веществ
в объектах окружающей среды, атмосферном
воздухе, воздухе рабочей зоны
и сельскохозяйственной продукции
Сборник
методических указаний
МУК
4.1.1960, 4.1.1961, 4.1.1963
- 4.1.1980-05
1. Разработаны Московской сельскохозяйственной академией им. К.А. Тимирязева, Учебно-научным консультационным центром «Токсикология пестицидов и агрохимикатов» (Калинин В.А., Калинина Т.С., Рыбакова О.И., Калинин А.В.).
2. Рекомендованы к утверждению Комиссией по санитарно-гигиеническому нормированию Минздрава России (протокол № 1 от 31 марта 2005 г.).
3. Утверждены Главным государственным санитарным врачом Российской Федерации, руководителем Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека Главным государственным санитарным врачом Российской Федерации Г.Г. Онищенко 21 апреля 2005 г.
4. Введены в действие с 1 июля 2005 г.
5. Введены впервые.
СОДЕРЖАНИЕ
|
УТВЕРЖДАЮ Руководитель
Федеральной службы ______________________ Г.Г. Онищенко 21 апреля 2005 г. Дата введения: 1 июля 2005 г. |
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение остаточных
количеств клопиралида
в семенах, масле и соломке льна, в семенах
и масле рапса методом газожидкостной хроматографии
Методические указания
МУК 4.1.1976-05
1. Вводная часть
1.1. Краткая характеристика
Действующее вещество (д.в.): клопиралид.
Название по номенклатуре ИЮПАК: 3,6-дихлорпиридин-2-карбоновая кислота.
Структурная формула:
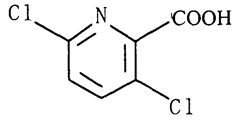
Эмпирическая формула: C6H3Cl2NO2.
Молекулярная масса: 192,0.
Химически чистое вещество: кристаллы белого цвета.
Температура плавления: 151 - 152 °С.
Растворимость (г/кг) при 20 °С: в воде - 1,0, циклогексаноне - 387, ацетоне - 153, ксилоле - 6,5.
В обычных условиях устойчив, с органическими и неорганическими основаниями образует хорошо растворимые в воде соли.
1.2. Краткая токсикологическая характеристика д.в.
LD50 для крыс - 4300 - 5000 мг/кг, LC50 (96 ч) для рыб - 103,5 мг/л. Нетоксичен для пчел и других насекомых.
Гигиенические нормативы: ВМДУ в семенах и масле рапса - 0,5 мг/кг.
1.3. Область применения препаратов
Гербицид для борьбы с некоторыми однолетними и многолетними двудольными сорными растениями.
2. Метод определения клопиралида в семенах, масле
и соломке льна, в семенах и масле рапса с применением
капиллярной газожидкостной хроматографии
2.1. Основные положения
2.1.1. Принцип метода
Метод основан на извлечении остаточных количеств клопиралида из анализируемого объекта органическими растворителями, проведении очистки экстракта перераспределением в системе несмешивающихся растворителей и метилировании клопиралида диазометаном.
Количественное определение проводят методом внешнего стандарта с применением капиллярной газожидкостной хроматографии и использованием детектора электронного захвата (ДЭЗ).
2.1.2. Избирательность метода
Метод специфичен в присутствии других применяемых пестицидов. Проведение очистки экстрактов, а также использование капиллярной колонки и селективного детектора позволяет устранять влияние коэкстрактивных веществ на результаты анализа.
2.1.3. Метрологическая характеристика метода
Диапазоны измеряемых концентраций, пределы обнаружения и другие метрологические параметры метода представлены в таблице.
Таблица
Метрологические параметры метода
|
Метрологические параметры |
Анализируемые объекты: |
||
|
семена льна и рапса |
масло льна и рапса |
соломка льна |
|
|
Предел обнаружения, мг/кг |
0,01 |
0,02 |
0,04 |
|
Диапазон определяемых концентраций, мг/кг |
0,01 - 0,08 |
0,02 - 0,16 |
0,04 - 0,32 |
|
Среднее значение определения, % |
80,0 |
77,4 |
80,7 |
|
Стандартное отклонение, % |
5,8 |
6,9 |
5,5 |
|
Относительное стандартное отклонение, % |
3,4 |
3,8 |
3,2 |
2.2. Реактивы, растворы, материалы
|
Аналитический стандарт клопиралида |
|
|
Азот газообразный вч |
ТУ 301-07-25-89 |
|
Ацетон, осч |
ТУ 2633-004-11291058-94 |
|
Ацетонитрил для хроматографии, хч |
ТУ 6-09-4326-76 |
|
Вата медицинская |
ТУ 9393-001-00302238-97 |
|
Вода дистиллированная и перегнанная над KMnO4 и щелочью |
|
|
н-Гексан, хч |
ТУ 6-09-3375-78 |
|
Дихлорметан, хч |
ТУ 6-09-2662-77 |
|
Изооктан эталонный |
|
|
Калия гидроокись, чда |
|
|
Натрий серно-кислый б/в (сульфат), чда |
|
|
Натрий хлористый, чда |
|
|
N-Нитрозометилмочевина, хч |
ТУ 6-09-11-1643-82 |
|
Серная кислота, осч |
|
|
Смесь н-гексан-этиловый эфир, 50:50, по объему |
|
|
Фильтры бумажные, «красная лента» |
ТУ 2642-001 -4262415 7-98 |
|
Фильтры бумажные, «белая лента» |
ТУ 2642-001-42624157-98 |
|
Фильтры бумажные, «синяя лента» |
ТУ 2642-001-42624157-98 |
|
Эфир этиловый (серный) |
ОСТ 84-2006-88 |
2.3. Приборы, аппаратура, посуда
|
Газовый хроматограф с ДЭЗ |
|
|
Колонка хроматографическая кварцевая капиллярная длиной 30 м, внутренним диаметром 0,32 мм с неподвижной фазой ВР-50, толщина пленки - 0,5 мкм |
|
|
Аппарат для встряхивания или аналогичный |
ТУ 64-1-1081-73 |
|
Весы аналитические типа ВЛА-200 |
ГОСТ 34104-80 |
|
Весы лабораторные типа ВЛКТ-500 |
ГОСТ 24104-80 |
|
Воронки делительные емкостью 250 и 500 мл |
|
|
Воронки химические конусные |
|
|
Индикаторная бумага универсальная |
ТУ 6-09-1181-76 |
|
Колбы-концентраторы емкостью 100 и 250 мл |
|
|
Колбы плоскодонные емкостью 100 и 300 мл |
|
|
Колбы мерные со шлифом емкостью 25, 50, 100 мл |
|
|
Колпачки алюминиевые для герметизации флаконов |
|
|
Мельница электрическая лабораторная или аналогичная |
ТУ 46-22-236-79 |
|
Микрошприц МШ-10 |
ТУ 2-833-106 |
|
Насос водоструйный |
ГОСТ 10696-75 |
|
Ротационный вакуумный испаритель типа ИР-1 или аналогичный |
|
|
Пипетки мерные емкостью 1, 2, 5 и 10 мл |
|
|
Приспособление для обжима колпачков на флаконах |
ТУ 42-2-2442-73 |
|
Пробирки мерные со шлифом емкостью 5,0 мл |
|
|
Стаканы химические на 100, 200 и 500 мл |
|
|
Установка для перегонки растворителей при атмосферном давлении |
|
|
Установка для упаривания растворителей в токе азота |
|
|
Установка ультразвуковая «Серьга» УЗМ002 или аналогичная |
|
|
Флаконы стеклянные (типа пенициллиновых) емкостью 5,0 мл |
ТУ 64-2-10-87 |
|
Электроплитка |
2.4. Подготовка к определению
2.4.1. Подготовка и очистка растворителей
Перед началом работы рекомендуется проверить чистоту применяемых органических растворителей. Для этого 100 мл растворителя упаривают в ротационном вакуумном испарителе при температуре 40 °С до объема 1,0 мл и хроматографируют. При обнаружении мешающих определению примесей очистку растворителей производят в соответствии с общепринятыми методиками.
2.4.2. Приготовление стандартных растворов
Основной стандартный раствор с содержанием 100 мкг/мл готовят растворением в ацетоне 0,01 г клопиралида в мерной колбе емкостью 100 мл. Раствор хранят в холодильнике при температуре 4 - 6 °С не более трех месяцев.
Рабочие стандартные растворы с концентрациями 1,6; 0,8; 0,4 и 0,2 мкг/мл готовят из основного стандартного раствора клопиралида последовательным разбавлением ацетоном. Рабочие растворы хранят в холодильнике при температуре 4 - 6 °С не более месяца.
В модельных опытах при изучении полноты извлечения используют аналогично приготовленные растворы клопиралида в гексане.
Для приготовления калибровочных растворов в мерные пробирки со шлифом емкостью 5,0 мл вносят по 1,0 мл рабочих растворов клопиралида с концентрациями 0,2; 0,4; 0,8 и 1,6 мкг/мл. Растворитель в пробирках упаривают в токе азота досуха и проводят метилирование клопиралида по п. 2.4.3.
2.4.3. Метилирование клопиралида
В пробирки с сухим остатком добавляют по 2,0 мл свежеприготовленного по п. 2.4.5 эфирного раствора диазометана. Пробирки закрывают пробками и ставят на 12 - 14 часов (на ночь) в холодильник с температурой 4 - 6 °С. После этого в пробирки добавляют по 1,0 мл изооктана и упаривают растворители в токе азота до 1,0 мл.
2.4.4. Построение калибровочного графика
Для построения калибровочного графика в инжектор хроматографа (п. 2.7.3) вводят по 1 мкл приготовленных по п. 2.4.3 растворов, содержащих клопиралид (в виде производного) в концентрациях 0,2; 0,4; 0,8 и 1,6 мкг/мл. Осуществляют не менее трех параллельных измерений и находят среднее значение высоты (площади) хроматографического пика для каждой концентрации. Строят калибровочный график зависимости высоты (площади) хроматографического пика в мм (мм2) от концентрации клопиралида в рабочем растворе в мкг/мл.
2.4.5. Приготовление эфирного раствора диазометана
(из расчета метилирования экстрактов 2 проб)
N-нитрозометилмочевину массой 0,5 г помещают во флакон емкостью 2,0 - 3,0 мл и герметизируют резиновой пробкой и колпачком с помощью приспособления для обжима колпачков на флаконах. Этиловый эфир объемом 4,0 мл вносят в другой флакон емкостью 5,0 мл, герметизируют резиновой пробкой и колпачком и охлаждают в морозильной камере холодильника в течение 30 мин.
После этого флаконы через предварительно проколотые пробки соединяют гибкой тефлоновой трубкой (внутр. диам. ~ 1,5 - 2,0 мм), одним концом погружая ее в этиловый эфир на всю глубину (флакон с охлажденным этиловым эфиром обязательно должен еще иметь свободный выход в атмосферу). Во флакон с нитрозометилмочевиной, используя шприц с тонкой иглой и прокалывая пробку, добавляют по каплям по стенке 50 % водный раствор гидроокиси калия (~ 0,3 мл) до прекращения реакции. Этиловый эфир при насыщении диазометаном окрашивается в ярко желтый цвет.
Внимание! Приготовление эфирного раствора диазометана и процедуру метилирования необходимо обязательно проводить в работающем вытяжном шкафу.
2.5. Отбор, первичная обработка и хранение проб
Отбор проб для анализа проводят в соответствии с «Унифицированными правилами отбора проб сельскохозяйственной продукции, продуктов питания и объектов окружающей среды для определения микроколичеств пестицидов», утвержденными 21.08.1979 г., № 2051-79.
Пробы семян просушивают до стандартной влажности и хранят при комнатной температуре в закрытой стеклянной или полиэтиленовой таре.
Пробы соломки просушивают при комнатной температуре до воздушно-сухого состояния и хранят в закрытой стеклянной или полиэтиленовой таре.
Пробы масла хранят в холодильнике при 4 - 6 °С в закрытой стеклянной таре.
2.6. Подготовка проб к определению
Пробы семян перед анализом рассыпают на бумаге или кальке и пинцетом удаляют включения. Семена измельчают на лабораторной мельнице и после перемешивания измельченной массы отбирают усредненную аналитическую пробу.
Пробы соломки измельчают и после перемешивания измельченной массы отбирают усредненную аналитическую пробу.
2.7. Проведение определения
2.7.1. Семена льна и рапса
2.7.1.1. Экстракция клопиралида и очистка экстракта
Аналитическую пробу семян массой 20 ± 0,1 г помещают в плоскодонную колбу емкостью 300 мл, добавляют 150 мл смеси ацетон-дистиллированная вода (90:10), слегка встряхивают и подвергают обработке ультразвуком в УЗ-бане в течение 10 мин. После этого содержимое колбы фильтруют через бумажный фильтр «красная лента» в колбу-концентратор емкостью 250 мл. Содержимое колбы с пробой промывают 50 мл смеси ацетон-дистиллированная вода (90:10), которую также фильтруют в колбу-концентратор.
При использовании аппарата для встряхивания в плоскодонную колбу с аналитической пробой вносят 150 мл смеси ацетон-дистиллированная вода (90:10) и встряхивают в течение 60 мин. После этого содержимое колбы фильтруют через бумажный фильтр «красная лента» в колбу-концентратор емкостью 250 мл. Содержимое колбы с пробой промывают 50 мл смеси ацетон-дистиллированная вода (90:10), которую также фильтруют в колбу-концентратор.
Колбу-концентратор с объединенным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворитель до объема 10 - 20 мл при температуре 40 °С. В колбу-концентратор добавляют 200 мл бидистиллированной воды, 2,0 мл 5,0 %-го водного раствора серной кислоты и содержимое колбы перемешивают встряхиванием. Колбу-концентратор помещают в холодильник и выдерживают при температуре 4 - 6 °С в течение 4 - 5 ч. После этого содержимое колбы фильтруют через бумажный фильтр «белая лента» в делительную воронку емкостью 500 мл. В воронку добавляют 10 % водный раствор гидроокиси калия до рН 9 - 10, 10 мл насыщенного водного раствора хлористого натрия и после перемешивания 75 мл дихлорметана. Содержимое воронки энергично встряхивают в течение 2 мин. После 15 минутного отстаивания нижний дихлорметановый слой сливают и отбрасывают. Процедуру очистки экстракта повторяют с использованием 50 мл дихлорметана. Далее в воронку добавляют 40 мл насыщенного водного раствора хлористого натрия и после перемешивания 75 мл н-гексана. Содержимое воронки энергично встряхивают в течение 2 мин. После 5 минутного отстаивания нижний водный слой сливают в химический стакан емкостью 500 мл, а верхний гексановый слой сливают и отбрасывают.
Водный раствор пробы, находящийся в химическом стакане, подкисляют концентрированной серной кислотой до рН 2,0 и переносят в чистую делительную воронку емкостью 500 мл. В воронку добавляют 75 мл смеси гексан-этиловый эфир (50:50) и встряхивают в течение 2 мин. После полного разделения нижний водный слой сливают в химический стакан, а верхний, гексано-эфирный, фильтруют через фильтр «синяя лента» со слоем безводного сульфата натрия (толщина - 1,0 - 1,5 см) в колбу-концентратор емкостью 150 мл. Экстрагирование и фильтрование повторяют с использованием 50 мл смеси гексан-этиловый эфир (50:50). Нижний водный слой отбрасывают.
Колбу-концентратор с объединенным гексано-эфирным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворители при температуре 40 °С до объема 3 - 5 мл. Остаток экстракта количественно переносят в мерную пробирку со шлифом емкостью 10 мл и упаривают растворители в токе азота досуха при температуре 40 °С.
2.7.1.2. Метилирование клопиралида
В пробирку с сухим остатком добавляют 2,0 мл свежеприготовленного по п. 2.4.5. эфирного раствора диазометана. Пробирку закрывают пробкой и ставят на 12 - 14 ч (на ночь) в холодильник с температурой 4 - 6 °С. После этого в пробирку добавляют 1,0 мл изооктана и упаривают растворители в токе азота до 1,0 мл. Газохроматографический анализ на содержание клопиралида проводят по п. 2.7.4.
2.7.2. Масло льна и рапса
2.7.2.1. Экстракция клопиралида и очистка экстракта
Пробу масла массой 10,0 ± 0,1 г растворяют в 50 мл н-гексана (насыщенного ацетонитрилом) в плоскодонной колбе емкостью 100 мл и после этого гексановый раствор масла переносят в делительную воронку емкостью 250 мл. Колбу промывают 50 мл ацетонитрила (насыщенного н-гексаном) и переносят его в воронку. Содержимое воронки встряхивают в течение 2 мин. После 5 минутного отстаивания нижний ацетонитрильный слой сливают в колбу-концентратор емкостью 100 мл.
Колбу промывают еще 25 мл ацетонитрила (насыщенного н-гекcаном) и также переносят в воронку (250 мл). Содержимое воронки встряхивают в течение 2 мин, отстаивают 5 мин и нижний ацетонитрильный слой объединяют с предыдущим. Верхний гексановый слой отбрасывают.
Колбу-концентратор с объединенным ацетонитрильным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворитель досуха при температуре 50 °С. Сухой остаток растворяют в 20 мл ацетона. К раствору добавляют 200 мл бидистиллированной воды, 2,0 мл 5,0 %-го водного раствора серной кислоты и содержимое колбы перемешивают встряхиванием. Колбу-концентратор помещают в холодильник и выдерживают при температуре 4 - 6 °С в течение 4 - 5 ч. После этого содержимое колбы фильтруют через бумажный фильтр белая лента в делительную воронку емкостью 500 мл. В воронку добавляют 10 %-й водный раствор гидроокиси калия до рН 9 - 10, 10 мл насыщенного водного раствора хлористого натрия и после перемешивания 75 мл дихлорметана. Содержимое воронки энергично встряхивают в течение 2 мин. После 15 минутного отстаивания нижний дихлорметановый слой сливают и отбрасывают. Процедуру очистки экстракта повторяют с использованием 50 мл дихлорметана. Далее в воронку добавляют 40 мл насыщенного водного раствора хлористого натрия и после перемешивания 75 мл н-гексана. Содержимое воронки энергично встряхивают в течение 2 мин. После 5 минутного отстаивания нижний водный слой сливают в химический стакан емкостью 500 мл, а верхний гексановый слой сливают и отбрасывают.
Водный раствор пробы, находящийся в химическом стакане, подкисляют концентрированной серной кислотой до рН 2,0 и переносят в чистую делительную воронку емкостью 500 мл. В воронку добавляют 75 мл смеси гексан-этиловый эфир (50:50) и встряхивают в течение 2 мин. После полного разделения нижний водный слой сливают в химический стакан, а верхний, гексано-эфирный, фильтруют через фильтр «синяя лента» со слоем безводного сульфата натрия (толщина - 1,0 - 1,5 см) в колбу-концентратор емкостью 150 мл. Экстрагирование и фильтрование повторяют с использованием 50 мл смеси гексан-этиловый эфир (50:50). Нижний водный слой отбрасывают.
Колбу-концентратор с объединенным гексано-эфирным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворители при температуре 40 °С до объема 3 - 5 мл. Остаток экстракта количественно переносят в мерную пробирку со шлифом емкостью 10 мл и упаривают растворители в токе азота досуха при температуре 40 °С.
2.7.2.2. Метилирование клопиралида
В пробирку с сухим остатком добавляют 2,0 мл свежеприготовленного по п. 2.4.5 эфирного раствора диазометана. Пробирку закрывают пробкой и ставят на 12 - 14 ч (на ночь) в холодильник с температурой 4 - 6 °С. После этого в пробирку добавляют 1,0 мл изооктана и упаривают растворители в токе азота до 1,0 мл. Газохроматографический анализ на содержание клопиралида проводят по п. 2.7.4.
2.7.3. Соломка льна
2.1.3.1. Экстракция клопиралида и очистка экстракта
Аналитическую пробу соломки массой 5,0 ± 0,1 г помещают в плоскодонную колбу емкостью 300 мл, добавляют 150 мл смеси ацетон-дистиллированная вода (80:20), слегка встряхивают и подвергают обработке ультразвуком в УЗ-бане в течение 10 мин. После этого содержимое колбы фильтруют через бумажный фильтр «красная лента» в колбу-концентратор емкостью 250 мл. Содержимое колбы с пробой промывают 50 мл смеси ацетон-дистиллированная вода (90:10), которую также фильтруют в колбу-концентратор.
При использовании аппарата для встряхивания в плоскодонную колбу с аналитической пробой вносят 150 мл смеси ацетон-дистиллированная вода (80:20) и встряхивают в течение 60 мин. После этого содержимое колбы фильтруют через бумажный фильтр «красная лента» в колбу-концентратор емкостью 250 мл. Содержимое колбы с пробой промывают 50 мл смеси ацетон-дистиллированная вода (90:10), которую также фильтруют в колбу-концентратор.
Колбу-концентратор с объединенным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворитель до объема 10 - 20 мл при температуре 40 °С. В колбу-концентратор добавляют 200 мл бидистиллированной воды, 2,0 мл 5,0 %-го водного раствора серной кислоты и содержимое колбы перемешивают встряхиванием. Колбу-концентратор помещают в холодильник и выдерживают при температуре 4 - 6 °С в течение 4 - 5 ч. После этого содержимое колбы фильтруют через бумажный фильтр «белая лента» в делительную воронку емкостью 500 мл. В воронку добавляют 10 %-й водный раствор гидроокиси калия до рН 9 - 10, 10 мл насыщенного водного раствора хлористого натрия и после перемешивания 75 мл дихлорметана. Содержимое воронки энергично встряхивают в течение 2 мин. После 15 минутного отстаивания нижний дихлорметановый слой сливают и отбрасывают. Процедуру очистки экстракта повторяют с использованием 50 мл дихлорметана. Далее в воронку добавляют 40 мл насыщенного водного раствора хлористого натрия и после перемешивания 75 мл н-гексана. Содержимое воронки энергично встряхивают в течение 2 мин. После 5 минутного отстаивания нижний водный слой сливают в химический стакан емкостью 500 мл, а верхний гексановый слой сливают и отбрасывают.
Водный раствор пробы, находящийся в химическом стакане, подкисляют концентрированной серной кислотой до рН 2,0 и переносят в чистую делительную воронку емкостью 500 мл. В воронку добавляют 75 мл смеси гексан-этиловый эфир (50:50) и встряхивают в течение 2 мин. После полного разделения нижний водный слой сливают в химический стакан, а верхний, гексано-эфирный, фильтруют через фильтр «синяя лента» со слоем безводного сульфата натрия (толщина - 1,0 - 1,5 см) в колбу-концентратор емкостью 150 мл. Экстрагирование и фильтрование повторяют с использованием 50 мл смеси гексан-этиловый эфир (50:50). Нижний водный слой отбрасывают.
Колбу-концентратор с объединенным гексано-эфирным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворители при температуре 40 °С до объема 3 - 5 мл. Остаток экстракта количественно переносят в мерную пробирку со шлифом емкостью 10 мл и упаривают растворители в токе азота досуха при температуре 40 °С.
2.7.3.2. Метилирование клопиралида
В пробирку с сухим остатком добавляют 2,0 мл свежеприготовленного по п. 2.4.5. эфирного раствора диазометана. Пробирку закрывают пробкой и ставят на 12 - 14 часов (на ночь) в холодильник с температурой 4 - 6 °С. После этого в пробирку добавляют 1,0 мл изооктана и упаривают растворители в токе азота до 1,0 мл. Газохроматографический анализ на содержание клопиралида проводят по п. 2.7.4.
2.7.4. Условия хроматографирования
Газовый хроматограф с ДЭЗ.
Колонка хроматографическая кварцевая капиллярная длиной 30 м, внутренним диаметром 0,32 мм с неподвижной фазой ВР-50 (OV-17, НР-17, DB-17), толщина слоя - 0,5 мкм.
Температура колонки: программирование от 100 °С (3 мин) до 280 °С (25 мин) со скоростью 8,0 °С/мин.
Температура испарителя: 240 °С.
Температура детектора: 300 °С.
Расход газов: газа-носителя (азот вч) - 1,5 см /мин, дополнительного газа (азот вч) к ДЭЗ - 40 см3/мин.
Объем вводимой пробы: 1 мкл.
Время удерживания клопиралида (в виде производного): 14,20 ± 0,05 мин.
Предел детектирования: 0,1 нг.
Линейный диапазон детектирования: 0,2 - 2,0 нг.
2.7.5. Обработка результатов анализа
Содержание клопиралида рассчитывают методом внешнего стандарта по формуле:
|
|
Х - содержание клопиралида в пробе, мг/кг;
H1 - высота (площадь) пика анализируемого вещества, мм (мм2);
P0 - высота (площадь) пика стандартного вещества, мм (мм2);
А - концентрация стандартного раствора клопиралида, мкг/мл;
V - объем экстракта, подготовленного для хроматографирования мл;
m - масса (г) аналитической пробы.
3. Требования техники безопасности
Необходимо соблюдать общепринятые правила техники безопасности при работе с органическими растворителями, токсичными веществами, электронагревательными приборами и сжатыми газами, а также требования, изложенные в документации к приборам.
4. Контроль погрешности измерений
Оперативный контроль погрешности и воспроизводимости результатов измерений осуществляется в соответствии с ГОСТ ИСО 5725-1 - 6.2002 «Точность (парвильность и прецизионность методов и результатов измерений».
5. Разработчики
П.А. Тарарин, Т.А. Маханькова, Л.В. Григорьева (ВНИИ защиты растений, Санкт-Петербург).