ФЕДЕРАЛЬНОЕ АГЕНТСТВО
ПО
ТЕХНИЧЕСКОМУ РЕГУЛИРОВАНИЮ И МЕТРОЛОГИИ
|
|
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ |
ГОСТ Р |

Изделия медицинские
СИСТЕМЫ МЕНЕДЖМЕНТА КАЧЕСТВА
Системные требования для целей регулирования
ISO 13485:2003
Medical devices -
Quality management systems -
System requirements for regulatory purposes
(IDT)
Москва
ИПК Издательство стандартов
2004
Предисловие
Задачи, основные принципы и правила проведения работ по государственной стандартизации в Российской Федерации установлены ГОСТ Р 1.0-92 «Государственная система стандартизации Российской Федерации. Основные положения» и ГОСТ Р 1.2-92 «Государственная система стандартизации Российской Федерации. Порядок разработки государственных стандартов»
Сведения о стандарте
1 ПОДГОТОВЛЕН Закрытым акционерным обществом «ВНИИМП-ВИТА» на основе собственного аутентичного перевода стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 «Управление качеством и соответствующие аспекты для медицинских изделий»
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 10 ноября 2004 г. № 66-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 13485:2003 «Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования» ISO 13485:2003 «Medical devices - Quality management systems - System requirements for regulatory purposes».
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты, сведения о которых приведены в дополнительном приложении С
5 ВЗАМЕН ГОСТ Р 51536-99, ГОСТ Р 51537-99
Введение
Международный стандарт ИСО 13485 подготовлен Техническим комитетом ИСО/ТК 210 «Управление качеством и соответствующие аспекты для медицинских изделий».
Настоящее издание отменяет и заменяет [1] и [2]. Организации, которые в прошлом применяли [2], могут применять настоящий стандарт, исключив некоторые требования в соответствии с пунктом 1.2 настоящего стандарта.
Настоящее издание ИСО 13485 имеет новый титульный лист и отражает требования к обеспечению качества продукции, требования потребителя и другие аспекты системы менеджмента качества.
Общие положения
Настоящий стандарт определяет требования к системе менеджмента качества, которые могут применяться организацией при проектировании, разработке, производстве, монтаже и обслуживании медицинских изделий, а также при проектировании, разработке и обеспечении связанных с ними услуг.
Настоящий стандарт может также применяться внутренними и внешними по отношению к организации заинтересованными сторонами, включая органы по сертификации, для оценки способности организации удовлетворять требования потребителя и требования, установленные в соответствии с нормативными документами (далее - установленные требования).
Информация, обозначенная как «Примечание», является методическим указанием по пониманию или разъяснению соответствующего требования.
Требования к системе менеджмента качества, установленные в настоящем стандарте, являются дополняющими по отношению к техническим требованиям к продукции.
Для создания системы менеджмента качества требуется стратегическое решение организации. На разработку и внедрение системы менеджмента качества организации влияют изменяющиеся потребности, конкретные цели, выпускаемая продукция, применяемые процессы, размер и структура организации. Настоящий стандарт не предполагает единообразия в структуре систем менеджмента качества или документации.
Существует большое разнообразие медицинских изделий; некоторые частные требования настоящего стандарта применимы только к тем категориям медицинских изделий, определения которых приведены в разделе 3.
Процессный подход
Настоящий стандарт основывается на процессном подходе к менеджменту качества.
Любая деятельность, имеющая входы и преобразующая их в выходы, может рассматриваться как процесс.
Для успешного функционирования организация должна определить множество взаимосвязанных процессов и управлять ими.
Часто выход одного процесса образует непосредственно вход следующего.
Применение в организации системы процессов наряду с их идентификацией и взаимодействием, а также менеджмент процессов, могут считаться процессным подходом.
Связь с ИСО 9001
Настоящий стандарт является автономным, но основывается на [3].
Пункты или подпункты настоящего стандарта, текст которых аутентичен тексту [3], набраны прямым шрифтом. То, что они представлены в неизменном виде, нашло отражение в приложении В.
Текст настоящего стандарта, не аутентичный тексту [3], или предложение или абзац, содержащие такой текст, набраны курсивом. Суть и причины изменений в тексте указаны в приложении В.
Связь с ИСО/ТО 14969
[4] представляет собой технический отчет, содержащий руководящие указания по внедрению настоящего стандарта.
Совместимость с другими системами менеджмента
Настоящий стандарт сохраняет структуру [3] для удобства пользователей медицинскими изделиями.
Настоящий стандарт не содержит специальных требований к другим системам менеджмента, например к частным системам менеджмента охраны окружающей среды, безопасности и профессионального здравоохранения, а также финансового менеджмента.
Однако настоящий стандарт позволяет организации согласовать или интегрировать свою собственную систему менеджмента качества с другими системами менеджмента с соответствующими требованиями. Организация может адаптировать действующую систему (системы) менеджмента для создания системы менеджмента качества, соответствующей требованиям настоящего стандарта.
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Изделия медицинские
СИСТЕМЫ МЕНЕДЖМЕНТА КАЧЕСТВА
Системные требования для целей регулирования
Medical devices.
Quality management systems.
System requirements for regulatory purposes
Дата введения - 2005-07-01
1 Область применения
1.1 Общие положения
Настоящий стандарт определяет требования к системе менеджмента качества в случаях, когда организации необходимо продемонстрировать способность поставлять медицинские изделия и предоставлять связанное с ними обслуживание, отвечающее требованиям потребителя и установленным требованиям, применимым к этим медицинским изделиям и сопутствующим услугам.
Цель настоящего стандарта - содействие внедрению в системы менеджмента качества гармонизированных установленных требований к медицинским изделиям. Настоящий стандарт включает в себя некоторые специальные требования к медицинским изделиям, а также исключает некоторые требования [3], которые не являются установленными, в связи с чем организации, чьи системы менеджмента качества отвечают требованиям настоящего стандарта, не могут заявлять о соответствии [3], пока их системы менеджмента качества не будут удовлетворять всем без исключения требованиям [3] (см. также приложение В).
1.2 Применение
Требования настоящего стандарта распространяются на организации, предлагающие на рынок медицинские изделия, независимо от вида или численности этих организаций.
Если установленные требования позволяют исключить управление проектированием и разработкой изделий (7.3), то это может служить основанием для исключения соответствующих требований из конкретной системы менеджмента качества. В этом случае для данной системы менеджмента качества должны быть сделаны альтернативные поправки. Ответственность за обеспечение соответствия исключения управления проектированием и разработкой изделий требованиям настоящего стандарта лежит на самой организации [4.2.2, перечисление а) и 7.3].
Если какое-либо требование (требования) раздела 7 настоящего стандарта нельзя применить ввиду специфики конкретного (конкретных) медицинского изделия (изделий), на которое (которые) распространяется система менеджмента качества, то организации не следует включать такое требование (требования) в свою систему менеджмента качества (4.2.2, перечисление а).
Ответственность за применение к медицинскому изделию (изделиям) процессов, соответствующих требованиям настоящего стандарта, но которые не осуществляются организацией, несет конкретная организация, и это следует учитывать в системе менеджмента качества этой организации (4.1, перечисление а).
В настоящем стандарте несколько раз встречаются понятия «если соответствует» и «когда соответствует». Если требование содержит одно из этих понятий, то оно считается «соответствующим», пока организация не предоставит документированное обоснование несоответствия настоящему стандарту. Требование считается «соответствующим», если его выполнение необходимо для того, чтобы:
а) изделие удовлетворяло установленным требованиям и/или
б) организация могла выполнять корректирующие действия.
2 Нормативные ссылки
В настоящем стандарте использована ссылка на следующий стандарт:
ИСО 9000-2000 Системы менеджмента качества. Основные положения и словарь (ISO 9000:2000 Quality management systems - Fundamentals and vocabulary)
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов по указателю «Национальные стандарты», составленному по состоянию на 1 января текущего года, и по соответствующим информационным указателям, опубликованным в текущем году. Если ссылочный документ заменен (изменен), то при пользовании настоящим стандартом следует руководствоваться замененным (измененным) стандартом. Если ссылочный документ отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены термины и определения в соответствии с ИСО 9000, а также следующие термины с соответствующими определениями.
Приведенные ниже термины, используемые в настоящем стандарте для описания цепи поставки, были изменены для отражения применяемого в настоящее время словаря терминов:
поставщик - организация - потребитель
Термин «организация» заменяет термин «поставщик», используемый в ИСО 9001:1996, и относится к объекту, в отношении которого применяется настоящий стандарт. Кроме того, термин «поставщик» заменяет термин «субподрядчик».
В тексте настоящего стандарта термин «продукция» может означать также «услугу».
Там, где требования определены как применимые к «медицинским изделиям», эти же требования равнозначно применимы и к связанным с ними услугам, предоставляемым той же организацией.
Термины и определения, помещенные ниже, следует рассматривать как наиболее общие, поскольку термины и определения, существующие в национальных нормативных документах, могут отличаться от них и быть предпочтительными.
3.1 активное имплантируемое медицинское изделие (active implantable medical device): Активное медицинское изделие, предназначенное для полного или частичного введения хирургическим или другим медицинским способом в тело человека либо, посредством медицинского вмешательства, в естественное отверстие и остающееся там после введения.
3.2 активное медицинское изделие (active medical device): Медицинское изделие, для функционирования которого необходим источник электрической или другой энергии, отличной от энергии, производимой телом человека или силой тяжести.
3.3 пояснительное уведомление (advisory notice): Уведомление, изданное организацией-поставщиком после поставки медицинского изделия, содержащее дополнительную информацию и/или рекомендации о том, какие корректирующие или предупреждающие действия должны быть предприняты при:
- применении медицинского изделия;
- модификации медицинского изделия;
- возврате медицинского изделия организации-поставщику;
- выходе из строя медицинского изделия.
Примечание - Пояснительное уведомление может потребоваться для приведения требований к медицинскому изделию в соответствие с национальными или региональными нормативными документами.
3.4 жалоба потребителя (customer complaint): Письменное, электронное или устное сообщение о недостатках, связанных с идентичностью, качеством, сроком службы, надежностью, безопасностью или функционированием медицинского изделия, введенного в обращение.
3.5 имплантируемое медицинское изделие (implantable medical device): Медицинское изделие, предназначенное для полного или частичного введения в тело человека или в естественное отверстие либо для замещения эпителиальной поверхности или поверхности глаза путем хирургического вмешательства на срок не менее 30 дней, которое может быть удалено только посредством хирургического либо другого медицинского вмешательства.
Примечание - Это определение применяется к имплантируемым медицинским изделиям, отличным от активных имплантируемых медицинских изделий.
3.6 маркировка (labelling): Информация в письменной, печатной или графической форме, нанесенная на медицинское изделие, тару, упаковку или сопровождающая медицинское изделие и связанная с его идентификацией, техническим описанием и применением, но не включенная в сопроводительные документы.
Примечание - В некоторых региональных или национальных нормативных документах термин «маркировка» интерпретируется как «информация, предоставляемая изготовителем».
3.7 медицинские изделия (изделия) (medical devices): Приборы, аппараты, инструменты, устройства, комплекты, комплексы, системы с программными средствами, оборудование, приспособления, перевязочные и шовные средства, стоматологические материалы, наборы реагентов, контрольные материалы и стандартные образцы, изделия из полимерных, резиновых и иных материалов, применяемые в медицинских целях по отдельности или в сочетании друг с другом и предназначенные изготовителем для:
- профилактики, диагностики, лечения заболеваний, проведения медицинских процедур, исследований медицинского характера, замены или модификации частей тела человека, восстановления или компенсации нарушенных или утраченных физиологических функций, контроля над зачатием;
- воздействия на организм человека, при котором их функциональное назначение не реализуется, но может поддерживаться химическим, фармакологическим, иммунологическим или метаболическим взаимодействием с организмом человека.
3.8 стерильное медицинское изделие (sterile medical device): Категория медицинских изделий, соответствующих требованиям к стерильности.
Примечание - Допускается требования к стерильности медицинских изделий устанавливать в национальных или региональных нормативных документах.
4 Система менеджмента качества
4.1 Общие требования
Организация должна разрабатывать, документировать, внедрять и поддерживать в рабочем состоянии систему менеджмента качества, а также поддерживать ее результативность в соответствии с требованиями настоящего стандарта.
Организация должна:
а) определять процессы, необходимые для системы менеджмента качества, и их применение во всей организации (1.2);
б) определять последовательность и взаимодействие этих процессов;
в) определять критерии и методы, необходимые для обеспечения результативности как при осуществлении, так и при управлении этими процессами;
г) обеспечивать наличие ресурсов и информации, необходимых для поддержки этих процессов и их мониторинга;
д) осуществлять мониторинг, измерение и анализ этих процессов;
е) выполнять действия, необходимые для достижения запланированных результатов и поддержания результативности этих процессов.
Организация должна осуществлять менеджмент этих процессов в соответствии с требованиями настоящего стандарта.
Если организация решает передать сторонним организациям выполнение какого-либо процесса, влияющего на соответствие продукции требованиям, она должна обеспечивать со своей стороны контроль такого процесса. Управление им должно быть определено в системе менеджмента качества (8.5.1).
Примечание - В процессы, необходимые для системы менеджмента качества, следует включать процессы управленческой деятельности руководства, обеспечения ресурсами, процессы жизненного цикла продукции и измерения.
4.2 Требования к документации
4.2.1 Общие положения
Документация системы менеджмента качества должна включать:
а) документально оформленные заявления о политике и целях в области качества;
б) руководство по качеству;
в) документированные процедуры, требуемые настоящим стандартом;
г) документы, необходимые организации для обеспечения эффективного планирования, осуществления процессов и управления ими;
д) записи, требуемые настоящим стандартом (4.2.4);
е) другие документы, указанные в национальных или региональных нормативных документах.
Там, где в настоящем стандарте указано, что требование, процедура, какая-либо деятельность или специальное мероприятие должны быть «документированы», подразумевается, что их также следует выполнять и поддерживать в рабочем состоянии.
Для каждого типа или модели медицинского изделия организация должна создать и поддерживать в рабочем состоянии файл, содержащий или идентифицирующий документы, определяющие спецификации на продукцию и требования к системе менеджмента качества (4.2.3). Эти документы должны также определять полный процесс изготовления и, если необходимо, монтажа и обслуживания медицинского изделия.
Примечания
1 Степень документирования системы менеджмента качества одной организации может отличаться от другой в зависимости от:
а) размера организации и вида деятельности;
б) сложности и взаимодействия процессов;
в) компетенции персонала.
2 Документация может быть в любой форме и на любом носителе.
4.2.2 Руководство по качеству
Организация должна разрабатывать и поддерживать в рабочем состоянии руководство по качеству, содержащее:
а) область применения системы менеджмента качества, включая подробности и обоснование любых исключений и/или неприменения (1.2);
б) документированные процедуры, разработанные для системы менеджмента качества, или ссылки на них;
в) описание взаимодействия процессов системы менеджмента качества.
Руководство по качеству должно определять структуру документов, используемых в системе менеджмента качества.
4.2.3 Управление документацией
Документами системы менеджмента качества необходимо управлять. Записи - специальный вид документов, и ими надо управлять согласно требованиям, приведенным в 4.2.4.
Для определения необходимых средств управления должна быть разработана документированная процедура, предусматривающая:
а) проверку и утверждение документов на адекватность до их выпуска;
б) анализ и актуализацию, по мере необходимости, и переутверждение документов;
в) обеспечение идентификации изменений и статуса пересмотра документов;
г) обеспечение наличия соответствующих версий документов в местах их применения;
д) обеспечение сохранения документов четкими и легко идентифицируемыми;
е) обеспечение идентификации документов внешнего происхождения и управление их рассылкой;
ж) предотвращение непреднамеренного использования устаревших документов и применение соответствующей идентификации таких документов, оставленных для каких-либо целей.
Организация должна обеспечивать анализ и утверждение изменений в документах либо должностным лицом, утверждавшим первоначальный документ, либо другим специально назначенным должностным лицом, имеющим доступ к соответствующей исходной информации, на основании которой принимается решение.
Организация должна определять период времени, в течение которого следует хранить не менее одной копии устаревших регулирующих документов. На этот период должен быть обеспечен доступ к документам, в соответствии с которыми медицинское изделие было изготовлено и испытано, в течение, по крайней мере, срока службы изделия, определенного организацией, но не менее срока хранения любой итоговой записи (4.2.4) или согласно установленным требованиям.
4.2.4 Управление записями
Записи должны вестись и поддерживаться в рабочем состоянии для предоставления свидетельств соответствия требованиям и результативности функционирования системы менеджмента качества. Они должны оставаться четкими, легко идентифицируемыми и восстанавливаемыми. Надо разработать документированную процедуру для определения средств управления, требуемых при идентификации, хранении, защите, восстановлении, определении сроков хранения и изъятии записей.
Организация должна хранить записи в течение, по крайней мере, срока службы медицинского изделия, определенного организацией, но не менее двух лет с момента выпуска изделия организацией или в соответствии с установленными требованиями.
5 Ответственность руководства
5.1 Обязательства руководства
Высшее руководство организации должно подтверждать свою приверженность разработке и внедрению системы менеджмента качества, а также поддержанию ее результативности посредством:
а) доведения до сведения организации важности выполнения требований потребителя, а также законодательных и обязательных требований;
б) разработки политики в области качества;
в) обеспечения разработки целей в области качества;
г) проведения анализа со стороны руководства;
д) обеспечения необходимыми ресурсами.
Примечание - В настоящем стандарте установленные требования ограничены только безопасностью и функционированием конкретного медицинского изделия.
5.2 Ориентация на потребителя
Высшее руководство организации должно обеспечивать определение и выполнение требований потребителя (7.2.1 и 8.2.1)
5.3 Политика в области качества
Высшее руководство должно обеспечивать, чтобы политика в области качества:
а) соответствовала целям организации;
б) включала обязательство соответствовать требованиям системы менеджмента качества и поддерживать ее результативность;
в) создавала основы для постановки и анализа целей в области качества;
г) была доведена до сведения персонала организации и понятна ему;
д) анализировалась на постоянную пригодность.
5.4 Планирование
5.4.1 Цели в области качества
Высшее руководство организации должно обеспечивать, чтобы цели в области качества, включая те, которые необходимы для выполнения требований к продукции (7.1, перечисление а), были установлены в соответствующих подразделениях и на соответствующих уровнях. Цели в области качества должны быть измеримыми и согласуемыми с политикой в области качества.
5.4.2 Планирование создания и развития системы менеджмента качества
Высшее руководство должно обеспечивать:
а) планирование создания и развития системы менеджмента качества для выполнения требований, приведенных в 4.1, а также для достижения целей в области качества;
б) сохранение целостности системы менеджмента качества при планировании и внедрении в нее изменений.
5.5 Ответственность, полномочия и обмен информацией
5.5.1 Ответственность и полномочия
Высшее руководство организации должно обеспечивать:
определение ответственности и полномочий, которые необходимо документировать, и доведение их до сведения персонала организации;
взаимодействие и независимость персонала, руководящего, выполняющего и верифицирующего работу по обеспечению качества, и определять полномочия, необходимые для выполнения этих задач.
Особое внимание следует уделить назначению конкретных лиц, ответственных за виды деятельности, связанные с наблюдением за эксплуатацией готовых изделий, а также оформлением отчета об инцидентах (8.2.1 и 8.5.2).
5.5.2 Представитель руководства
Высшее руководство должно назначить представителя из состава руководства, который независимо от других обязанностей должен нести ответственность и иметь полномочия, распространяющиеся на:
а) обеспечение разработки, внедрения и поддержания в рабочем состоянии процессов, требуемых системой менеджмента качества;
б) представление отчетов высшему руководству о функционировании системы менеджмента качества и необходимости улучшения (8.5);
в) обеспечение внутри организации понимания установленных требований и требований потребителя.
Примечание - В ответственность представителя руководства может быть включено поддержание связи с внешними сторонами по вопросам, касающимся системы менеджмента качества
5.5.3 Внутренний обмен информацией
Высшее руководство должно обеспечивать разработку в организации соответствующих процессов обмена информацией, в том числе по вопросам результативности системы менеджмента качества.
5.6 Анализ со стороны руководства
5.6.1 Общие положения
Высшее руководство должно анализировать через запланированные интервалы систему менеджмента качества организации с целью обеспечения ее постоянной пригодности, адекватности и результативности. В анализ следует включать оценку возможностей улучшения и потребности в изменениях в системе менеджмента качества организации, в том числе в политике и целях в области качества.
Записи об анализе со стороны руководства должны поддерживаться в рабочем состоянии (4.2.4).
5.6.2 Входные данные для анализа
Входные данные для анализа со стороны руководства должны включать следующую информацию:
а) результаты аудитов (проверок);
б) обратную связь с потребителями;
в) функционирование процессов и соответствие продукции;
г) статус предупреждающих и корректирующих действий;
д) последующие действия, вытекающие из предыдущего анализа со стороны руководства;
е) изменения, которые могли бы повлиять на систему менеджмента качества;
ж) рекомендации по улучшению;
з) новые или пересмотренные установленные требования.
5.6.3 Выходные данные для анализа
Выходные данные анализа со стороны руководства должны включать все решения и действия, относящиеся к:
а) улучшениям, необходимым для поддержания результативности системы менеджмента качества и ее процессов;
б) улучшению продукции согласно требованиям потребителей;
в) потребности в ресурсах.
6 Менеджмент ресурсов
6.1 Обеспечение ресурсами
Организация должна определять и обеспечивать ресурсы, требуемые для:
а) внедрения системы менеджмента качества и поддержания ее результативности;
б) удовлетворения установленных требований и требований потребителя.
6.2 Человеческие ресурсы
6.2.1 Общие положения
Персонал, выполняющий работу, влияющую на качество продукции, должен быть компетентным в соответствии с полученным образованием, подготовкой, навыками и опытом.
6.2.2 Компетентность, осведомленность и подготовка
Организация должна:
а) определять необходимую компетентность персонала, выполняющего работу, которая влияет на качество продукции;
б) обеспечивать подготовку или предпринимать другие действия с целью удовлетворения этих потребностей;
в) оценивать результативность предпринятых мер;
г) обеспечивать осведомленность своего персонала об актуальности и важности его деятельности и вкладе в достижение целей в области качества;
д) поддерживать в рабочем состоянии соответствующие записи об образовании, подготовке, навыках и опыте (4.2.4).
Примечание - Национальные или региональные нормативные документы могут содержать требования к конкретной организации о разработке документированных процедур для определения необходимости подготовки персонала.
6.3 Инфраструктура
Организация должна определять, обеспечивать и поддерживать в рабочем состоянии инфраструктуру, необходимую для достижения соответствия требованиям к продукции. Инфраструктура может включать:
а) здания, рабочее пространство и связанные с ним средства труда;
б) оборудование для процессов (как технические, так и программные средства);
в) службы обеспечения (например транспорт или связь).
Организация должна разработать документированные требования к действиям, поддерживающим ее инфраструктуру, включая частоту их проведения, если эти действия или их отсутствие могут повлиять на качество продукции.
Записи об этих действиях должны поддерживаться в рабочем состоянии (4.2.4).
6.4 Производственная среда
Организация должна создавать производственную среду, необходимую для достижения соответствия требованиям к продукции, и управлять ею.
Это включает следующие требования:
а) организация должна разработать документированные требования к состоянию здоровья, гигиене и одежде персонала, если контакт между персоналом и продукцией или производственной средой может отрицательно повлиять на качество продукции (7.5.1.2.1);
б) если производственная среда может отрицательно сказаться на качестве продукции, организация должна разработать документированные требования к производственной среде и документированные процедуры или производственные инструкции для мониторинга и контроля производственной среды (7.5.1.2.1);
в) организация должна гарантировать соответствующую подготовку персонала, временно работающего в особых условиях производственной среды, или его нахождение под наблюдением подготовленного лица (6.2.2, перечисление б);
г) при необходимости разработку и документирование специальных мер для контроля загрязненной или потенциально загрязненной продукции с целью предотвращения загрязнения остальной продукции, производственной среды или загрязнения персонала (7.5.3.1).
7 Процессы жизненного цикла продукции
7.1 Планирование процессов жизненного цикла продукции
Организация должна планировать и разрабатывать процессы, необходимые для обеспечения жизненного цикла продукции. Планирование процессов жизненного цикла продукции должно быть согласовано с требованиями к другим процессам системы менеджмента качества (4.1).
При планировании процессов жизненного цикла продукции организация должна установить, если это целесообразно:
а) цели в области качества и требования к продукции;
б) потребность в разработке процессов, документов, а также в обеспечении ресурсами для конкретной продукции;
в) необходимую деятельность по верификации и валидации, мониторингу, контролю и испытаниям для конкретной продукции, а также критерии приемки продукции;
г) записи, необходимые для обеспечения свидетельства того, что процессы жизненного цикла продукции и произведенная продукция соответствуют требованиям (4.2.4).
Результаты этого планирования должны быть в форме, соответствующей практике организации.
Организация должна разработать документированные требования к управлению риском на всех этапах жизненного цикла продукции. Записи по управлению риском должны поддерживаться в рабочем состоянии (4.2.4 и примечание 3).
Примечания
1 Документ, определяющий процессы системы менеджмента качества (включая процессы жизненного цикла продукции) и ресурсы, которые предстоит применять к конкретной продукции, проекту или контракту, может рассматриваться как план качества.
2 При разработке процессов жизненного цикла продукции организация может также применять требования 7.3.
3 См. [5] по управлению риском.
7.2 Процессы, связанные с потребителями
7.2.1 Определение требований, относящихся к продукции
Организация должна определить:
а) требования потребителя, включая требования к поставке и деятельности после поставки;
б) требования, не определенные потребителем, но необходимые для конкретного или предполагаемого использования, когда оно известно;
в) законодательные и другие обязательные требования, относящиеся к продукции;
г) любые дополнительные требования, определенные организацией.
7.2.2 Анализ требований, относящихся к продукции
Организация должна анализировать требования, относящиеся к продукции. Этот анализ должен проводиться до принятия организацией обязательства поставлять продукцию потребителю (например участия в тендерах, принятия контрактов или заказов, принятия изменений к контрактам или заказам) и должен обеспечивать:
а) определение и документирование требований к продукции;
б) согласование требований контракта или заказа, отличающихся от ранее сформулированных;
в) способность организации удовлетворять определенные требования.
Записи результатов анализа и последующих действий, вытекающих из анализа, должны поддерживаться в рабочем состоянии (4.2.4).
Если потребители не выдвигают документированных требований, организация должна подтвердить их у потребителя до принятия к исполнению.
Если требования к продукции изменены, организация должна обеспечить, чтобы соответствующие документы были исправлены, а заинтересованный персонал был поставлен в известность об изменившихся требованиях.
Примечание - В некоторых ситуациях, таких как продажи, осуществляемые через Интернет, практически нецелесообразно проводить официальный анализ каждого заказа. Вместо этого анализ может распространяться на соответствующую информацию о продукции, такую как каталоги или рекламные материалы.
7.2.3 Связь с потребителями
Организация должна определять и осуществлять эффективные меры по поддержанию связи с потребителями, касающиеся:
а) информации о продукции;
б) прохождения запросов, контракта или заказа, включая поправки;
в) обратной связи с потребителем, включая жалобы потребителей (8.2.1);
г) пояснительных уведомлений (8.5.1).
7.3 Проектирование и разработка
7.3.1 Планирование проектирования и разработки
Организация должна разрабатывать документированные процедуры проектирования и разработки медицинских изделий.
Организация обязана планировать и управлять проектированием и разработкой продукции.
В ходе планирования проектирования и разработки организация должна устанавливать:
а) стадии проектирования и разработки;
б) виды деятельности по анализу, верификации, валидации и передаче проекта (см. примечание), соответствующие каждой стадии проектирования и разработки;
в) ответственность и полномочия в области проектирования и разработки.
Организация должна управлять взаимодействием различных групп, занятых проектированием и разработкой, с целью обеспечения эффективной связи и четкого распределения ответственности.
Результаты планирования должны быть документированы и, если необходимо, актуализированы при совершенствовании проектирования и разработки (4.2.3).
Примечание - Деятельность по передаче проекта в процессе проектирования и разработки медицинского изделия должна гарантировать, что выходные данные проектирования и разработки будут верифицированы как соответствующие требованиям к процессу изготовления, прежде чем станут окончательными спецификациями продукции.
7.3.2 Входные данные для проектирования и разработки
Входные данные, относящиеся к требованиям к продукции, должны быть определены, а записи должны поддерживаться в рабочем состоянии (4.2.4).
Входные данные должны включать:
а) функциональные и эксплуатационные требования, требования к безопасности согласно назначению;
б) соответствующие законодательные и другие обязательные требования;
в) там, где это целесообразно, информацию, взятую из предыдущих аналогичных проектов;
г) другие требования, важные для проектирования и разработки;
д) выходные данные по управлению риском (7.1).
Входные данные должны быть проанализированы на соответствие установленным требованиям и утверждены.
Требования должны быть полными, недвусмысленными и непротиворечивыми.
7.3.3 Выходные данные проектирования и разработки
Выходные данные проектирования и разработки должны быть представлены в форме, позволяющей провести верификацию относительно входных требований к проектированию и разработке, а также должны быть утверждены до их последующего использования.
Выходные данные проектирования и разработки должны:
а) соответствовать входным требованиям к проектированию и разработке;
б) обеспечивать соответствующей информацией по закупкам, производству и обслуживанию;
в) содержать критерии приемки продукции или ссылки на них;
г) определять характеристики продукции, существенные для ее безопасного и правильного использования.
Записи выходных данных проектирования и разработки должны поддерживаться в рабочем состоянии (4.2.4).
Примечание - Записи выходных данных проектирования и разработки могут включать спецификации, производственные процессы, технические чертежи и журналы технических записей или исследований.
7.3.4 Анализ проекта и разработки
На тех стадиях, где это целесообразно, должен проводиться систематический анализ проекта и разработки в соответствии с запланированными мероприятиями (7.3.1) с целью:
а) оценки способности результатов проектирования и разработки удовлетворять требованиям;
б) выявления любых проблем и внесения предложений по необходимым действиям.
Такой анализ должны выполнять представители служб, имеющих отношение к анализируемым стадиям проекта и разработки, а также другие специалисты организации (5.5.1 и 6.2.1).
Записи результатов анализа и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4).
7.3.5 Верификация проекта и разработки
Верификация должна осуществляться в соответствии с запланированными мероприятиями (7.3.1), чтобы удостовериться, что выходные данные проектирования и разработки соответствуют входным требованиям. Записи результатов верификации и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4).
7.3.6 Валидация проекта и разработки
Валидация проекта и разработки должна проводиться в соответствии с запланированными мероприятиями (7.3.1) для обеспечения соответствия готовой продукции требованиям к ее назначению или специальному применению. Валидация должна быть проведена до поставки продукции или введения ее в эксплуатацию (см. примечание 1).
Записи результатов валидации и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4).
Организация обязана проводить клинические и/или эксплуатационные испытания медицинских изделий как часть мероприятий по валидации проекта и разработки в соответствии с требованиями национальных или региональных нормативных документов (см. примечание 2).
Примечания
1 Если медицинское изделие подлежит валидации только после сборки и монтажа на месте его применения, поставка продукции не считается законченной до тех пор, пока продукция не будет формально передана потребителю.
2 Представление медицинского изделия для проведения клинических и/или эксплуатационных испытаний не считается поставкой.
7.3.7 Управление изменениями проекта и разработки
Изменения проекта и разработки должны быть идентифицированы, а записи должны поддерживаться в рабочем состоянии. Изменения должны быть проанализированы, верифицированы и подтверждены соответствующим образом, а также согласованы до внесения. Анализ изменений проекта и разработки должен включать оценку влияния изменений на составные части изделий и уже поставленную продукцию.
Записи результатов анализа изменений и любых необходимых действий должны поддерживаться в рабочем состоянии (4.2.4).
7.4 Закупки
7.4.1 Процесс закупок
Организация должна разработать документированные процедуры, обеспечивающие соответствие закупленной продукции установленным требованиям к закупкам.
Тип и степень управления, применяемые по отношению к поставщику и закупленной продукции, должны зависеть от ее воздействия на последующие стадии жизненного цикла продукции или готовую продукцию.
Организация должна оценивать и выбирать поставщиков на основе их способности поставлять продукцию в соответствии с требованиями организации. Должны быть разработаны критерии отбора, оценки и повторной оценки. Записи результатов оценивания и любых необходимых действий, вытекающих из оценки, должны поддерживаться в рабочем состоянии (4.2.4).
7.4.2 Информация по закупкам
Информация по закупкам должна описывать заказанную продукцию, включая, где это необходимо:
а) требования к утверждению продукции, процедур, процессов и оборудования;
б) требования к квалификации персонала;
в) требования к системе менеджмента качества.
Организация должна обеспечивать адекватность установленных требований к закупкам до их сообщения поставщику.
В той степени, в какой это необходимо для осуществления прослеживаемости (7.5.3.2), организация должна поддерживать в рабочем состоянии соответствующую информацию по закупкам, то есть документы (4.2.3) и записи (4.2.4).
7.4.3 Верификация закупленной продукции
Организация должна разработать и осуществлять контроль или другую деятельность, необходимую для обеспечения соответствия закупленной продукции установленным требованиям к закупкам.
Если организация или потребитель ее продукции предлагают осуществить верификацию на предприятии поставщика, то организация должна установить в информации по закупкам предполагаемые меры по проверке и порядок выпуска продукции у поставщика.
Записи по верификации должны поддерживаться в рабочем состоянии (4.2.4).
7.5 Производство и обслуживание
7.5.1 Управление производством и обслуживанием
7.5.1.1 Общие требования
Организация должна планировать и обеспечивать производство и обслуживание в управляемых условиях. Управляемые условия должны включать, если это целесообразно:
а) наличие информации, описывающей характеристики продукции;
б) наличие документированных процедур, рабочих инструкций, эталонных процедур измерения и, если необходимо, справочного материала;
в) применение подходящего оборудования;
г) наличие и применение контрольных и измерительных приборов;
д) проведение мониторинга и измерений;
е) осуществление выпуска, поставки и действий после поставки продукции;
ж) выполнение конкретных действий по маркировке и упаковке.
Организация должна разработать форму записей и поддерживать их в рабочем состоянии (4.2.4) для каждой партии медицинских изделий, чтобы обеспечить их прослеживаемость в соответствии с 7.5.3 и установить количество произведенной продукции и продукции, предназначенной для продажи. Записи по каждой партии изделий должны быть верифицированы и утверждены.
Примечание - Партией может считаться одно медицинское изделие.
7.5.1.2 Управление производством и обслуживанием. Специальные требования
7.5.1.2.1 Чистота продукции и контроль загрязненности
Организация должна разработать документированные требования к чистоте продукции, если:
а) перед стерилизацией и/или применением продукция проходит очистку в организации;
б) продукция поставляется в нестерильном виде и подлежит очистке перед стерилизацией и/или применением;
в) продукция поставляется в нестерильном виде и ее чистота не имеет значения для применения;
г) реагенты для очистки продукции должны быть удалены при ее изготовлении.
Если продукция подвергается очистке в соответствии с перечислениями а) или б), требования, содержащиеся в (6.4, перечисление а) и (6.4, перечисление б), перед процедурой очистки не применяются.
7.5.1.2.2 Работы по монтажу
При необходимости организация должна разработать документированные требования, содержащие критерии приемки и верификации монтажа медицинского изделия.
Если согласованные с потребителем требования позволяют выполнять монтаж не только силами организации или ее полномочного представителя, организация должна разработать документированные требования к такому монтажу и его верификации.
Записи по монтажу и верификации, которые осуществляет организация или ее полномочный представитель, должны поддерживаться в рабочем состоянии (4.2.4).
7.5.1.2.3 Деятельность по обслуживанию
Если требование к обслуживанию является специальным, организация должна, при необходимости, разработать документированные процедуры, рабочие инструкции, справочные материалы и эталонные процедуры измерения для осуществления обслуживания и его верификации в соответствии со специальным требованием.
Записи по обслуживанию, выполняемому организацией, должны поддерживаться в рабочем состоянии (4.2.4).
Примечание - Обслуживание может включать, например, ремонт и техническое обслуживание.
7.5.1.3 Специальные требования к стерильным медицинским изделиям
Организация должна поддерживать записи по параметрам процессов стерилизации, применяемых для каждой партии стерилизуемой продукции (4.2.4). Записи по стерилизации должны прослеживаться для каждой партии произведенных медицинских изделий (7.5.1.1).
7.5.2 Валидация процессов производства и обслуживания
7.5.2.1 Общие требования
Организация должна подтверждать все процессы производства и обслуживания, результаты которых нельзя проверить посредством последовательного мониторинга или измерения. К ним относятся все процессы, недостатки которых становятся очевидными только после начала использования продукции.
Валидация должна продемонстрировать способность этих процессов достигать запланированных результатов.
Организация должна разработать меры по этим процессам, включая, если это приемлемо:
а) определенные критерии для анализа и утверждения процессов;
б) утверждение соответствующего оборудования и квалификации персонала;
в) применение конкретных методов и процедур;
г) требования к записям (4.2.4);
д) повторную валидацию.
Организация должна разрабатывать документированные процедуры валидации применения компьютерного программного обеспечения (и изменений такого обеспечения и/или его применения) при производстве и обслуживании, которые могут оказывать влияние на способность продукции удовлетворять установленным требованиям. Такое программное обеспечение следует утверждать до его первого применения.
Записи о результатах валидации должны поддерживаться в рабочем состоянии (4.2.4).
7.5.2.2 Специальные требования к стерильным медицинским изделиям
Организация должна разрабатывать документированные процедуры валидации процессов стерилизации. Процессы стерилизации следует утверждать до их первого применения.
Записи о результатах валидации процессов стерилизации должны поддерживаться в рабочем состоянии (4.2.4).
7.5.3 Идентификация и прослеживаемость
7.5.3.1 Идентификация
Организация должна идентифицировать продукцию на протяжении всего жизненного цикла с помощью соответствующих средств и разрабатывать документированные процедуры для такой идентификации продукции.
Организация должна разрабатывать документированные процедуры, гарантирующие, что медицинские изделия, возвращенные в организацию как несоответствующие, идентифицированы и отделены от изделий, соответствующих установленным требованиям (6.4, перечисление г).
7.5.3.2 Прослеживаемость
7.5.3.2.1 Общие положения
Организация должна разрабатывать документированные процедуры прослеживаемости. Такие процедуры должны определять степень прослеживаемости продукции и необходимые записи (4.2.4, 8.3 и 8.5).
Если прослеживаемость является требованием, то организация должна управлять специальной идентификацией продукции и регистрировать ее (4.2.4).
Примечание - Управление конфигурацией продукции является средством, с помощью которого можно осуществлять идентификацию и прослеживаемость.
7.5.3.2.2 Специальные требования к активным имплантируемым медицинским изделиям и имплантируемым медицинским изделиям
В записи, необходимые для осуществления прослеживаемости, организация должна включать сведения обо всех компонентах, материалах и условиях окружающей среды, если они могут явиться причиной того, что медицинское изделие не удовлетворяет установленным к нему требованиям.
Организация должна требовать от своих представителей или дистрибьюторов поддерживать записи о распределении медицинских изделий для достижения прослеживаемости и доступности этих записей для контроля.
Организация должна обеспечивать регистрацию грузополучателя (наименования и адреса) (4.2.4).
7.5.3.3 Идентификация статуса
Организация должна идентифицировать статус продукции по отношению к требованиям мониторинга и измерений.
Идентификация статуса продукции должна поддерживаться на всех этапах ее производства, хранения, монтажа и обслуживания для обеспечения отправления, применения или монтажа только продукции, прошедшей все необходимые виды контроля и испытаний (или имеющей официальное разрешение на отклонение от установленных требований).
7.5.4 Собственность потребителей
Организация должна проявлять заботу о собственности потребителя, пока она находится под управлением организации или используется ею. Организация должна идентифицировать, верифицировать, защищать и сохранять собственность потребителя, предоставленную для использования или включения в продукцию. Если собственность потребителя утеряна, повреждена или признана непригодной для использования, потребитель должен быть об этом извещен, а записи должны поддерживаться в рабочем состоянии (4.2.4).
Примечание - Собственностью потребителя может быть интеллектуальная собственность или конфиденциальная информация о состоянии его здоровья.
7.5.5 Сохранение соответствия продукции
Организация должна разрабатывать документированные процедуры или рабочие инструкции по сохранению соответствия продукции установленным требованиям при осуществлении технологических процессов внутри организации и доставке продукции к месту назначения.
Сохранение соответствия должно включать в себя идентификацию, погрузочно-разгрузочные работы, упаковку, хранение и защиту. Сохранение должно также применяться и к составным частям продукции.
Организация должна разработать документированные процедуры или рабочие инструкции по управлению продукцией с ограниченным сроком хранения либо продукцией, требующей специальных условий хранения. Такие специальные условия хранения должны регистрироваться и быть управляемыми.
7.6 Управление устройствами для мониторинга и измерений
Организация должна определить мониторинг и измерения, которые предстоит осуществлять, а также устройства для мониторинга и измерения, необходимые для обеспечения свидетельства соответствия продукции установленным требованиям (7.2.1).
Организация должна разработать документированные процедуры обеспечения проведения мониторинга и измерений в соответствии с установленными требованиями.
Там, где необходимо обеспечивать имеющие законную силу результаты, измерительное оборудование должно быть:
а) откалибровано и проверено в установленные периоды или перед его применением по образцовым эталонам, передающим размеры единиц в сравнении с международными или национальными эталонами. При отсутствии таких эталонов база, использованная для калибровки или поверки, должна быть зарегистрирована;
б) отрегулировано или повторно отрегулировано по мере необходимости;
в) идентифицировано с целью установления статуса калибровки;
г) защищено от регулировок, которые сделали бы недействительными результаты измерения;
д) защищено от повреждения и ухудшения состояния в ходе обращения, технического обслуживания и хранения.
Кроме того, организация должна оценить и зарегистрировать правомочность предыдущих результатов измерения, если обнаружено, что оборудование не соответствует требованиям. Организация должна предпринять соответствующее действие в отношении такого оборудования и любой измеренной продукции. Записи результатов калибровки и поверки должны поддерживаться в рабочем состоянии (4.2.4).
Если при мониторинге и измерении установленных требований используют компьютерные программные средства, их способность удовлетворять предполагаемому применению должна быть подтверждена. Это должно быть осуществлено до начала применения и повторно подтверждено по мере необходимости.
Примечание - См. руководящие указания в [6].
8 Измерение (оценка), анализ и улучшение
8.1 Общие положения
Организация должна планировать и применять процессы мониторинга, измерения (оценки), анализа и улучшения, необходимые для:
а) демонстрации соответствия продукции;
б) обеспечения соответствия системы менеджмента качества;
в) поддержания результативности системы менеджмента качества.
Это должно включать определение применяемых методов, в том числе статистических, и область их использования.
Примечание - Национальные или региональные нормативные документы могут содержать требования к разработке документированных процедур для внедрения статистических методов и управления их применением.
8.2 Мониторинг и измерение (оценка)
8.2.1 Обратная связь
В качестве одного из способов оценки функционирования системы менеджмента качества организация должна проводить мониторинг информации, относящейся к удовлетворению организацией требований потребителя.
Должны быть установлены методы получения и использования этой информации.
Организация должна разработать документированную процедуру по системе обратной связи (7.2.3, перечисление в) с целью получения раннего предупреждения о проблемах в области качества и введения в процессы корректирующих и предупреждающих действий (8.5.2 и 8.5.3).
Если национальные или региональные нормативные документы требуют от организации накопления опыта на основании информации, полученной после продажи медицинского изделия, анализ такого опыта может стать частью системы обратной связи (8.5.1).
8.2.2 Внутренние аудиты (проверки)
Организация должна проводить внутренние аудиты (проверки) через запланированные интервалы с целью установления того, что система менеджмента качества:
а) соответствует запланированным мероприятиям (7.1), требованиям настоящего стандарта и требованиям к системе менеджмента качества, разработанным организацией;
б) внедрена результативно и поддерживается в рабочем состоянии.
Программа аудитов (проверок) должна планироваться с учетом статуса и важности процессов и участков, подлежащих аудиту, а также результатов предыдущих аудитов. Критерии, область применения, частота и методы аудитов должны быть определены. Выбор аудиторов и проведение аудитов должны обеспечивать объективность и беспристрастность процесса аудита. Аудиторы не должны проверять свою собственную работу.
Ответственность и требования к планированию и проведению аудитов, а также к отчету о результатах и поддержанию записей в рабочем состоянии (4.2.4) должны быть определены в документированной процедуре.
Руководство, ответственное за проверяемые области деятельности, должно обеспечивать, чтобы действия предпринимались без излишней отсрочки для устранения обнаруженных несоответствий и вызвавших их причин. Последующие действия должны включать верификацию предпринятых мер и отчет о результатах верификации (8.5.2).
Примечание - См. [7] - руководство по аудиту.
8.2.3 Мониторинг и измерение (оценка) процессов
Организация должна применять подходящие методы мониторинга и, где это целесообразно, измерения (оценки) процессов системы менеджмента качества. Эти методы должны демонстрировать способность процессов достигать запланированных результатов. Если запланированные результаты не достигнуты, то, когда это целесообразно, должны предприниматься коррекции и корректирующие действия для обеспечения соответствия продукции.
8.2.4 Мониторинг и измерение (оценка) продукции
8.2.4.1 Общие требования
Организация должна проводить мониторинг и измерение характеристик продукции с целью верификации соответствия установленным требованиям. Мониторинг и измерение характеристик продукции должны быть проведены на соответствующих стадиях процессов жизненного цикла продукции и в соответствии с запланированными мероприятиями (7.1) и документированными процедурами (7.5.1.1).
Свидетельства соответствия критериям приемки должны поддерживаться в рабочем состоянии. Записи должны указывать лицо (а), санкционировавшее (ие) выпуск продукции (4.2.4).
Выпуск продукции и обслуживание не могут осуществляться до успешного завершения запланированных мероприятий (7.1).
8.2.4.2 Специальное требование для активных имплантируемых медицинских изделий и имплантируемых медицинских изделий
Организация должна вести записи по идентификации персонала (4.2.4), проводящего любые виды контроля или испытаний.
8.3 Управление несоответствующей продукцией
Организация должна обеспечивать, чтобы продукция, которая не соответствует требованиям, была идентифицирована и управлялась с целью предотвращения непреднамеренного использования или поставки. Средства управления, соответствующая ответственность и полномочия для работы с несоответствующей продукцией должны быть определены в документированной процедуре.
Организация должна решать вопрос о несоответствующей продукции одним или несколькими следующими способами:
а) осуществлять действия с целью устранения обнаруженного несоответствия;
б) санкционировать применение, выпуск или приемку продукции под разрешение на отклонение от установленных требований;
в) осуществлять действия с целью предотвращения ее первоначального предполагаемого использования или применения.
Организация должна гарантировать, что несоответствующая продукция будет принята при получении разрешения на отклонение от установленных требований только в том случае, если это допускается установленными требованиями. Записи об идентификации лица (лиц), имеющего (их) полномочия на выдачу разрешений на отклонение, должны поддерживаться в рабочем состоянии (4.2.4).
Записи о характере несоответствий и любых последующих предпринятых действиях, включая приемку продукции под разрешение, должны поддерживаться в рабочем состоянии (4.2.4).
Когда несоответствующая продукция исправлена, она должна быть подвергнута повторной верификации для подтверждения соответствия требованиям.
Если несоответствующая продукция выявлена после поставки или начала использования, организация должна предпринять действия, адекватные последствиям (или потенциальным последствиям) несоответствия.
Если продукция нуждается в переработке (один или более раз), организация обязана документировать процесс переработки в рабочей инструкции, на которую следует получить официальное разрешение и одобрение так же, как и на первоначальную рабочую инструкцию. Перед получением официального разрешения и одобрения должны быть определены и документированы (4.2.3 и 7.5.1) любые неблагоприятные последствия переработки продукции.
8.4 Анализ данных
Организация должна разработать документированную процедуру определения, сбора и анализа данных, необходимых для демонстрации результативности системы менеджмента качества и ее соответствия установленным требованиям, а также для оценки возможности повышения ее результативности.
Данные должны включать информацию, полученную в результате мониторинга и измерения также и из других соответствующих источников.
Анализ данных должен предоставлять информацию по:
а) обратной связи (8.2.1);
б) соответствию требованиям к продукции (7.2.1);
в) характеристикам и тенденциям процессов и продукции, включая возможности проведения предупреждающих действий;
г) поставщикам.
Записи результатов анализа данных должны поддерживаться в рабочем состоянии (4.2.4).
8.5 Улучшение
8.5.1 Общие положения
Организация должна идентифицировать и выполнять любые изменения, необходимые для обеспечения и поддержания постоянного соответствия установленным требованиям и результативности системы менеджмента качества, используя политику в области качества, устанавливая цели в этой области, а также используя результаты аудитов, анализа данных, корректирующих и предупреждающих действий и анализа со стороны руководства.
Организация должна установить документированные процедуры выпуска и применения пояснительных уведомлений. Следует обеспечить возможность проведения таких процедур в любое время.
Записи об изучении жалоб потребителей должны поддерживаться в рабочем состоянии (4.2.4). Если в ходе такого изучения будет установлено, что появлению жалоб потребителей способствует внешняя деятельность организации, между заинтересованными организациями следует провести обмен соответствующей информацией (4.1).
Если по любой жалобе потребителя не предприняты корректирующие и/или предупреждающие действия, причину следует объяснить (5.5.1) и зарегистрировать (4.2.4).
Если этого требуют национальные или региональные нормативные документы, организация должна разработать документированные процедуры уведомления регулирующих органов о тех инцидентах, которые соответствуют критериям к отчетности.
8.5.2 Корректирующие действия
Организация должна предпринимать корректирующие действия с целью устранения причин несоответствий для предупреждения повторного их возникновения. Корректирующие действия должны быть адекватными последствиям выявленных несоответствий.
Должна быть разработана документированная процедура для определения требований к:
а) анализу несоответствий (включая жалобы потребителей);
б) установлению причин несоответствий;
в) оцениванию необходимости действий, чтобы избежать повторения несоответствий;
г) определению и выполнению необходимых действий, включая, если целесообразно, обновление документации (4.2);
д) регистрации результатов любых расследований и принятых мер (4.2.4);
е) анализу предпринятых корректирующих действий и их результативности.
8.5.3 Предупреждающие действия
Организация должна определить действия с целью устранения причин потенциальных несоответствий для предупреждения их появления. Предупреждающие действия должны соответствовать возможным последствиям потенциальных проблем.
Должна быть разработана документированная процедура для определения требований к:
а) установлению потенциальных несоответствий и их причин;
б) оцениванию необходимости действий с целью предупреждения появления несоответствий;
в) определению и осуществлению необходимых действий;
г) регистрации результатов любых расследований и принятых мер (4.2.4);
д) анализу предпринятых предупреждающих действий и их результативности.
Приложение А
(справочное)
Соответствие между ИСО 13485:2003 и ИСО 13485:1996
Таблица А.1 - Соответствие между ИСО 13485:1996 и ИСО 13485:2003
|
ИСО 13485:1996 |
ИСО 13485:2003 |
|
1 Область применения |
|
|
2 Нормативные ссылки |
|
|
3 Определения |
|
|
4 Требования к системе качества (только заголовок) |
- |
|
4.1 Ответственность руководства (только заголовок) |
- |
|
4.1.1 Политика в области качества |
|
|
4.1.2 Организация (только заголовок) |
- |
|
4.1.2.1 Ответственность и полномочия |
|
|
4.1.2.2 Ресурсы |
|
|
4.1.2.3 Представитель руководства |
|
|
4.1.3 Анализ со стороны руководства |
|
|
4.2 Система качества (только заголовок) |
- |
|
4.2.1 Общие положения |
|
|
4.2.2 Процедуры системы качества |
|
|
4.2.3 Планирование качества |
|
|
4.3 Анализ контракта (только заголовок) |
- |
|
4.3.1 Общие положения (только заголовок) |
- |
|
4.3.2 Анализ |
|
|
4.3.3 Изменения к контракту |
|
|
4.3.4 Записи |
|
|
4.4 Управление проектированием (только заголовок) |
- |
|
4.4.1 Общие положения (только заголовок) |
- |
|
4.4.2 Планирование проектирования и разработки |
|
|
4.4.3 Организационное и техническое взаимодействие |
|
|
4.4.4 Входные проектные данные |
|
|
4.4.5 Выходные проектные данные |
|
|
4.4.6 Анализ проекта |
|
|
4.4.7 Проверка проекта |
|
|
4.4.8 Утверждение проекта |
|
|
4.4.9 Изменения проекта |
|
|
4.5 Управление документацией и данными (только заголовок) |
- |
|
4.5.1 Общие положения |
|
|
4.5.2 Утверждение и выпуск документации и данных |
|
|
4.5.3 Изменение документов и данных |
|
|
4.6 Закупки (только заголовок) |
- |
|
4.6.1 Общие положения (только заголовок) |
- |
|
4.6.2 Оценка субподрядчиков |
|
|
4.6.3 Информация по закупкам |
|
|
4.6.4 Проверка закупленной продукции |
|
|
4.7 Управление продукцией, поставляемой потребителем |
|
|
4.8 Идентификация и прослеживаемость продукции |
|
|
4.9 Управление процессами |
|
|
4.10 Контроль и испытания (только заголовок) |
- |
|
4.10.1 Общие положения |
|
|
4.10.2 Входной контроль и испытания |
|
|
4.10.3 Контроль и испытания в процессе производства |
|
|
4.10.4 Окончательный контроль и испытания |
|
|
4.10.5 Регистрация данных контроля и испытаний |
|
|
4.11 Управление контрольным, измерительным и испытательным оборудованием (только заголовок) |
- |
|
4.11.1 Общие положения |
|
|
4.11.2 Процедуры управления |
|
|
4.12 Статус контроля и испытаний |
|
|
4.13 Управление несоответствующей продукцией (только заголовок) |
- |
|
4.13.1 Общие положения |
|
|
4.13.2 Анализ и утилизация несоответствующей продукции |
|
|
4.14 Корректирующие и предупреждающие действия (только заголовок) |
- |
|
4.14.1 Общие положения |
|
|
4.14.2 Корректирующие действия |
|
|
4.14.3 Предупреждающие действия |
|
|
4.15 Погрузочно-разгрузочные работы, хранение, упаковка, консервация и поставка (только заголовок) |
- |
|
4.15.1 Общие положения |
|
|
4.15.2 Погрузочно-разгрузочные работы |
|
|
4.15.3 Хранение |
|
|
4.15.4 Упаковка |
|
|
4.15.5 Консервация |
|
|
4.15.6 Поставка |
|
|
4.16 Управление регистрацией данных о качестве |
|
|
4.17 Внутренние проверки качества |
|
|
4.18 Подготовка кадров |
|
|
4.19 Обслуживание |
|
|
4.20 Статистические методы (только заголовок) |
- |
|
4.20.1 Определение потребностей |
|
|
4.20.2 Процедуры |
Таблица А.2 - Соответствие между ИСО 13485:2003 и ИСО 13485:1996
|
ИСО 13485:2003 |
ИСО 13485:1996 |
|
1 Область применения |
1 |
|
1.1 Общие положения |
- |
|
1.2 Применение |
- |
|
2 Нормативные ссылки |
2 |
|
3 Термины и определения |
3 |
|
4 Система менеджмента качества (только заголовок) |
- |
|
4.1 Общие требования |
4.2.1 |
|
4.2 Требования к документации (только заголовок) |
- |
|
4.2.1 Общие положения |
4.2.2 |
|
4.2.2 Руководство по качеству |
4.2.1 |
|
4.2.3 Управление документацией |
4.5.1, 4.5.2, 4.5.3 |
|
4.2.4 Управление записями |
4.16 |
|
5 Ответственность руководства (только заголовок) |
- |
|
5.1 Обязательства руководства |
4.1.1 |
|
5.2 Ориентация на потребителя |
4.3.2 |
|
5.3 Политика в области качества |
4.1.1 |
|
5.4 Планирование (только заголовок) |
- |
|
5.4.1 Цели в области качества |
4.1.1 |
|
5.4.2 Планирование создания и развития системы менеджмента качества |
4.2.3 |
|
5.5 Ответственность, полномочия и обмен информацией (только заголовок) |
- |
|
5.5.1 Ответственность и полномочия |
4.1.2.1 |
|
5.5.2 Представитель руководства |
4.1.2.3 |
|
5.5.3 Внутренний обмен информацией |
- |
|
5.6 Анализ со стороны руководства (только заголовок) |
- |
|
5.6.1 Общие положения |
4.1.3 |
|
5.6.2 Входные данные для анализа |
- |
|
5.6.3 Выходные данные анализа |
- |
|
6 Менеджмент ресурсов (только заголовок) |
- |
|
6.1 Обеспечение ресурсами |
4.1.2.2 |
|
6.2 Человеческие ресурсы (только заголовок) |
- |
|
6.2.1 Общие положения |
4.1.2.2 |
|
6.2.2 Компетентность, осведомленность и подготовка |
4.18 |
|
6.3 Инфраструктура |
4.9 |
|
6.4 Производственная среда |
4.9, 4.15.1 |
|
7 Процессы жизненного цикла продукции (только заголовок) |
- |
|
7.1 Планирование процессов жизненного цикла продукции |
4.2.3, 4.10.1 |
|
7.2 Процессы, связанные с потребителями (только заголовок) |
- |
|
7.2.1 Определение требований, относящихся к продукции |
4.3.2, 4.4.4 |
|
7.2.2 Анализ требований, относящихся к продукции |
4.3.2, 4.3.3, 4.3.4 |
|
7.2.3 Связь с потребителями |
4.3.2 |
|
7.3 Проектирование и разработка (только заголовок) |
- |
|
7.3.1 Планирование проектирования и разработки |
4.4.2, 4.4.3 |
|
7.3.2 Входные данные для проектирования и разработки |
4.4.4 |
|
7.3.3 Выходные данные проектирования и разработки |
4.4.5 |
|
7.3.4 Анализ проекта и разработки |
4.4.6 |
|
7.3.5 Верификация проекта и разработки |
4.4.7 |
|
7.3.6 Валидация проекта и разработки |
4.4.8 |
|
7.3.7 Управление изменениями проекта и разработки |
4.4.9 |
|
7.4 Закупки (только заголовок) |
- |
|
7.4.1 Процесс закупок |
4.6.2 |
|
7.4.2 Информация по закупкам |
4.6.3 |
|
7.4.3 Верификация закупленной продукции |
4.6.4, 4.10.2 |
|
7.5 Производство и обслуживание (только заголовок) |
- |
|
7.5.1 Управление производством и обслуживанием |
4.9, 4.15.6, 4.19 |
|
7.5.2 Валидация процессов производства и обслуживания |
4.9 |
|
7.5.3 Идентификация и прослеживаемость |
4.8, 4.10.5, 4.12 |
|
7.5.4 Собственность потребителей |
4.7 |
|
7.5.5 Сохранение соответствия продукции |
4.15.2, 4.15.3, 4.15.4, 4.15.5 |
|
7.6 Управление устройствами для мониторинга и измерений |
4.10, 4.20.1, 4.20.2 |
|
8 Измерение (оценка), анализ и улучшение (только заголовок) |
- |
|
8.1 Общие положения |
4.11.1, 4.11.2 |
|
8.2 Мониторинг и измерения (оценка) (только заголовок) |
- |
|
8.2.1 Обратная связь |
- |
|
8.2.2 Внутренние аудиты (проверки) |
4.17 |
|
8.2.3 Мониторинг и измерение (оценка) процессов |
4.17, 4.20.1, 4.20.2 |
|
8.2.4 Мониторинг и измерение (оценка) продукции |
4.10.2, 4.10.3, 4.10.4, 4.10.5, 4.20.1, 4.20.2 |
|
8.3 Управление несоответствующей продукцией |
4.13.1, 4.13.2 |
|
8.4 Анализ данных |
4.20.1, 4.20.2 |
|
8.5 Улучшение (только заголовок) |
- |
|
8.5.1 Общие положения |
4.1.3 |
|
8.5.2 Корректирующие действия |
4.14.1, 4.14.2 |
|
8.5.3 Предупреждающие действия |
4.14.1, 4.14.3 |
Приложение В
(справочное)
Объяснение различий между ИСО 13485:2003 и ИСО 9001:2000
Настоящее приложение подтверждает сходство и различие как содержащих требования, так и содержащих информацию пунктов и подпунктов ИСО 13485:2003 и [3]. Приводятся также причины различий между ИСО 13485:2003 и [3]:
а) пункты или подпункты ИСО 13485:2003, аутентичные [3], заключены в таблице В.1 в квадратные скобки;
б) пункты или подпункты [3], измененные в ИСО 13485:2003 путем добавления к ним информации или посредством согласования с нормативными документами, касающимися медицинских изделий, приведены в левой колонке таблицы В.1. Соответствующие пункты или подпункты ИСО 13485:2003 приведены в правой колонке таблицы В.1;
в) пункты или подпункты [3], отсутствующие в ИСО 13485:2003 или измененные посредством удаления или существенного изменения требования, в первоначальном и неизменном виде приведены в левой колонке таблицы В.1. Соответствующие пункты или подпункты ИСО 13485:2003 и причины их изменения по сравнению [3] приведены в правой колонке таблицы В.1;
г) причины различий между ИСО 13485:2003 и [3] приведены в правой колонке таблицы В.1. Там, где различий в каком-либо пункте или подпункте нет, разница в тексте между ИСО 13485:2003 и ИСО 9001:2000 отражает задачу ИСО 13485:2003, состоящую в ознакомлении с действующими в мире нормативными документами и облегчении всемирной гармонизации новых нормативных документов на медицинские изделия.
Таблица В.1 - Объяснение различий между ИСО 9001:2000 и ИСО 13485:2003
|
ИСО 9001:2000 |
ИСО 13485:2003 |
|
Введение |
Введение |
|
Общие положения |
Общие положения |
|
Для создания системы менеджмента качества требуется стратегическое решение организации. На разработку и внедрение системы менеджмента качества организации влияют изменяющиеся потребности, конкретные цели, выпускаемая продукция, применяемые процессы, размер и структура организации. Настоящий стандарт не предполагает единообразия в структуре систем менеджмента качества или документации. Требования к системе менеджмента качества, установленные в настоящем стандарте, являются дополняющими по отношению к требованиям к продукции. Информация, обозначенная как «Примечание», является методическим указанием по пониманию или разъяснению соответствующего требования. Настоящий стандарт может использоваться внутренними и внешними сторонами, включая органы по сертификации, с целью оценки способности организации выполнять требования потребителей, регламентов и собственные требования. При разработке настоящего стандарта были учтены принципы менеджмента качества, установленные в ИСО 9000 и [8] |
Настоящий стандарт определяет требования к системе менеджмента качества, которые могут применяться организацией при проектировании, разработке, производстве, монтаже и обслуживании медицинских изделий, а также при проектировании, разработке и обеспечении связанных с ними услуг. Настоящий стандарт может также применяться внутренними и внешними по отношению к организации заинтересованными сторонами, включая органы по сертификации, для оценки способности организации удовлетворять требования потребителя и установленные требования. Информация, обозначенная как «Примечание», является методическим указанием по пониманию или разъяснению соответствующего требования. Требования к системе менеджмента качества, установленные в настоящем стандарте, являются дополняющими по отношению к техническим требованиям к продукции. Для создания системы менеджмента качества требуется стратегическое решение организации. На разработку и внедрение системы менеджмента качества организации влияют изменяющиеся потребности, конкретные цели, выпускаемая продукция, применяемые процессы, размер и структура организации. Настоящий стандарт не предполагает единообразия в структуре систем менеджмента качества или документации. Существует большое разнообразие медицинских изделий; некоторые частные требования настоящего стандарта применимы только к тем категориям медицинских изделий, определения которых приведены в разделе 3. Причина различий: кроме содержания раздела 4 любые изменения внесены в [3] только с целью возможности его применения в отношении медицинских изделий |
|
Процессный подход Настоящий стандарт направлен на применение процессного подхода при разработке, внедрении и улучшении результативности системы менеджмента качества с целью повышения удовлетворенности потребителей путем выполнения их требований. Для успешного функционирования организация должна определять и осуществлять менеджмент многочисленных взаимосвязанных видов деятельности. Деятельность, использующая ресурсы и управляемая с целью преобразования входов в выходы, может рассматриваться как процесс. Часто выход одного процесса образует непосредственно вход следующего. Применение в организации системы процессов наряду с их идентификацией и взаимодействием, а также менеджмент процессов, могут считаться процессным подходом. Преимущество процессного подхода состоит в непрерывности управления, которое он обеспечивает на стыке отдельных процессов в рамках их системы, а также при их комбинации и взаимодействии. При применении в системе менеджмента качества такой подход подчеркивает важность: а) понимания и выполнения требований; б) необходимости рассмотрения процессов с точки зрения добавленной ценности; в) достижения результатов выполнения процессов и их результативности; г) постоянного улучшения процессов, основанного на объективном измерении. Приведенная на рисунке 1 модель системы менеджмента качества, основанная на процессном подходе, иллюстрирует связи между процессами, представленными в разделах 4 - 8. Эта модель показывает, что потребители играют существенную роль при определении входных данных. Мониторинг удовлетворенности потребителей требует оценки информации о восприятии потребителями выполнения их требований. Приведенная на рисунке 1 модель охватывает все основные требования настоящего стандарта, не детализируя их. Примечание - Кроме того, ко всем процессам может применяться цикл «Plan - Do - Check - Act» (PDCA). Цикл PDCA можно кратко описать так: - планирование (plan) - разработайте цели и процессы, необходимые для достижения результатов в соответствии с требованиями потребителей и политикой организации; - осуществление (do) - внедрите процессы; - проверка (check) - постоянно контролируйте и измеряйте процессы и продукцию в сравнении с политикой, целями и требованиями на продукцию и сообщайте о результатах; - действие (act) - предпринимайте действия по постоянному улучшению показателей процессов.
Условные обозначения:
Рисунок 1 - Модель системы менеджмента качества, основанной на процессном подходе |
Процессный подход Настоящий стандарт основывается на процессном подходе к менеджменту качества. Любая деятельность, имеющая входы и преобразующая их в выходы, может рассматриваться как процесс. Для успешного функционирования организация должна определить множество взаимосвязанных процессов и управлять ими. Часто выход одного процесса образует непосредственно вход следующего. Применение в организации системы процессов наряду с их идентификацией и взаимодействием, а также менеджмент процессов, могут считаться процессным подходом. Причина различий. Большинство положений [3] предназначены для включения в технический отчет [4] - документ, обеспечивающий управление внедрением требований ИСО 13485:2003. Эта информация включена в [3], так как новая редакция [4] еще не существует |
|
Связь с ИСО 9004:2000 [3] и [8] были разработаны как согласованная пара стандартов на системы менеджмента качества для дополнения друг друга, но их можно применять также независимо. Несмотря на то, что у стандартов различные области применения, они имеют аналогичную структуру в целях создания условий для их использования как согласованной пары. [3] устанавливает требования к системе менеджмента качества, которые могут использоваться для внутреннего применения организациями в целях сертификации или заключения контрактов. Он направлен на результативность системы менеджмента качества при выполнении требований потребителей. |
Связь с ИСО 9001:2000 Настоящий стандарт является автономным, но основывается на [3]. Пункты или подпункты, текст которых аутентичен тексту [3], набраны прямым шрифтом. То, что они представлены в неизменном виде, нашло отражение в приложении В. Текст настоящего стандарта, не аутентичный [3], или предложение или абзац, содержащие такой текст, набраны курсивом. Суть и причины изменений в тексте указаны в приложении В. Связь с ИСО/ТО 14969 [4] представляет собой технический отчет, содержащий руководящие указания по внедрению настоящего стандарта. |
|
[8] содержит рекомендации по более широкому спектру целей системы менеджмента качества, чем [3], особенно по постоянному улучшению деятельности организации, а также ее эффективности и результативности. [8] рекомендуется как руководство для организаций, высшее руководство которых, преследуя цель постоянного улучшения деятельности, желает выйти за рамки требований [3]. Однако он не предназначен для целей сертификации или заключения контрактов |
Причина различий: между ИСО 13485:2003 и [8] отсутствует тесная связь. ИСО 13485:2003 связан главным образом с [3], а также с [4] |
|
Совместимость с другими системами менеджмента Настоящий стандарт согласован с [9] для улучшения совместимости этих двух стандартов в интересах сообщества пользователей. |
Совместимость с другими системами менеджмента Настоящий стандарт сохраняет структуру [3] для удобства пользователей медицинскими изделиями. |
|
Настоящий стандарт не содержит конкретных требований к другим системам менеджмента, таким как менеджмент охраны окружающей среды, менеджмент профессионального здоровья и безопасности, финансовый менеджмент или менеджмент рисков. Однако он позволяет организации согласовать или интегрировать свою собственную систему менеджмента качества с другими системами менеджмента с соответствующими требованиями. Организация может адаптировать действующую систему (системы) менеджмента для создания системы менеджмента качества, соответствующей требованиям настоящего стандарта |
Настоящий стандарт не содержит специальных требований к другим системам менеджмента, например к частным системам менеджмента охраны окружающей среды, безопасности и профессионального здравоохранения, а также финансового менеджмента. Однако настоящий стандарт позволяет организации согласовать или интегрировать свою собственную систему менеджмента качества с другими системами менеджмента с соответствующими требованиями. Организация может адаптировать действующую систему (системы) менеджмента для создания системы менеджмента качества, соответствующей требованиям настоящего стандарта. Причины различий: в этом пункте подчеркнута связь с [3]. Управление риском является ключевым требованием для многих видов деятельности и требований, связанных с системами менеджмента качества, для организаций, производящих медицинские изделия |
|
1 Область применения 1.1 Общие положения Настоящий стандарт устанавливает требования к системе менеджмента качества в тех случаях когда организация: а) нуждается в демонстрации своей способности поставлять продукцию, отвечающую требованиям потребителей и соответствующим обязательным требованиям; б) ставит своей целью повышение удовлетворенности потребителей посредством эффективного применения системы, включая процессы постоянного ее улучшения и обеспечения соответствия требованиям потребителей и обязательным требованиям. Примечание - В настоящем стандарте термин «продукция» применим только к предназначаемой для потребителя или затребованной им продукции |
1 Область применения 1.1 Общие положения Настоящий стандарт определяет требования к системе менеджмента качества в случаях, когда организации необходимо продемонстрировать способность поставлять медицинские изделия и предоставлять связанное с ними обслуживание, отвечающее требованиям потребителя и установленным требованиям, применимым к этим медицинским изделиям и сопутствующим услугам. Цель настоящего стандарта - содействие внедрению в системы менеджмента качества гармонизированных установленных требований к медицинским изделиям. Настоящий стандарт включает в себя некоторые специальные требования к медицинским изделиям, а также исключает некоторые требования [3], которые не являются установленными, в связи с чем организации, чьи системы менеджмента качества отвечают требованиям настоящего стандарта, не могут заявлять о соответствии [3], пока их системы менеджмента качества не будут удовлетворять всем без исключения требованиям [3], (см. приложение В). Причины различий: в пункте 1.1 применяются и разъясняются термины, имеющие отношение к медицинским изделиям. Кроме того, исключены термины «удовлетворенность потребителя» и «постоянное улучшение», поскольку они не соответствуют стандарту, цель которого заключается в облегчении всемирной гармонизации требований, установленных к системам менеджмента качества медицинских изделий. Во втором абзаце содержится разъяснение того, что целью ИСО 13485:2003 является облегчение всемирной гармонизации требований, установленных к системам менеджмента качества. Указано, что для этого требуется наличие некоторых дополнительных требований, отсутствующих в [3], и исключение других требований, имеющихся в [3]; кроме того, разъясняется, что соблюдение требований ИСО 13485:2003 не означает непременно соблюдение требований [3]. Термин «сопутствующее обслуживание» дважды добавляется к термину «медицинские изделия», поскольку определение «медицинские изделия» не включает понятие «обслуживание». Это противоречит [3], где понятие «обслуживание» является частью определения термина «продукция» |
|
1.2 Применение Требования настоящего стандарта предназначены для всех организаций, независимо от вида, размера и поставляемой продукции. Если какое-либо требование(я) настоящего стандарта нельзя применить ввиду специфики организации или ее продукции, допускается его исключение. При сделанных исключениях заявления о соответствии настоящему стандарту приемлемы, если эти исключения подпадают под требования, приведенные в разделе 7, и не влияют на способность или ответственность организации обеспечивать продукцией отвечающей требованиям потребителей и соответствующим обязательным требованиям |
1.2 Применение Требования настоящего стандарта распространяются на организации, предлагающие на рынок медицинские изделия, независимо от вида или численности этих организаций. Если установленные требования позволяют исключить управление проектированием и разработкой изделий (7.3), то это может служить основанием для исключения соответствующих требований из конкретной системы менеджмента качества. В этом случае для данной системы менеджмента качества должны быть сделаны альтернативные поправки. Ответственность за обеспечение соответствия исключения управления проектированием и разработкой изделий требованиям настоящего стандарта лежит на самой организации [4.2.2, перечисление а) и 7.3]. Если какое-либо требование (требования) раздела 7 настоящего стандарта нельзя применить ввиду специфики конкретного (конкретных) медицинского изделия (изделий), на которое (которые) распространяется система менеджмента качества, то организации не следует включать такое требование (требования) в свою систему менеджмента качества (4.2.2, перечисление а). Ответственность за применение к медицинскому изделию (изделиям) процессов, соответствующих требованиям настоящего стандарта, но которые не осуществляются организацией, несет конкретная организация, и это следует учитывать в системе менеджмента качества этой организации (4.1, перечисление а). В настоящем стандарте несколько раз встречаются понятия «если соответствует» и «когда соответствует». Если требование содержит одно из этих понятий, то оно считается «соответствующим», пока организация не предоставит документированное обоснование несоответствия настоящему стандарту. Требование считается «соответствующим», если его выполнение необходимо для того, чтобы: а) изделие удовлетворяло установленным требованиям и/или б) организация могла выполнять корректирующие действия. Причины различий: пункт 1.2 содержит разъяснение того, что требования ИСО 13485:2003 относятся именно к медицинским изделиям. Кроме того, усилена взаимосвязь между исключениями, относящимися к проектированию и разработке, что может иметь особое значение для требований, содержащихся в некоторых национальных нормативных документах. Наконец, объясняются различия между требованиями раздела 7, которые организация при наличии законных оснований может исключить из своей системы менеджмента качества (см. ограничения к 7.3), даже если она осуществляет виды деятельности, к которым относятся эти требования, и требованиями, которые организация может при наличии обоснования не включать в свою систему менеджмента качества, так как они относятся к видам деятельности, которые эта организация не осуществляет |
|
2 Нормативные ссылки Указанный ниже нормативный документ содержит положения, которые посредством ссылок в этом тексте составляют положения настоящего стандарта. Для датированных ссылок не применяются последующие изменения или пересмотры изданных документов. Однако участникам соглашений, основанных на настоящем стандарте, рекомендуется исследовать возможности применения последней редакции указанного ниже нормативного документа. Для недатированных ссылок применяется последнее издание нормативного документа. Члены ИСО и МЭК поддерживают в рабочем состоянии официальные списки действующих международных стандартов. ИСО 9000:2000 Системы менеджмента качества. Основные положения и словарь |
2 Нормативные ссылки В настоящем стандарте использованы ссылки на следующий стандарт: ИСО 9000-2000 Системы менеджмента качества. Основные положения и словарь (ISO 9000:2000 Quality management systems - Fundamentals and vocabulary) Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов (и классификаторов) на территории государства по соответствующему указателю стандартов (и классификаторов), составленному по состоянию на 1 января текущего года, и по соответствующим информационным указателям, опубликованным в текущем году. Если ссылочный документ заменен (изменен), то при пользовании настоящим стандартом следует руководствоваться замененным (измененным) стандартом. Если ссылочный документ отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку. |
|
3 Термины и определения В настоящем стандарте применяют термины и определения, данные в ИСО 9000:2000. Приведенные ниже термины, используемые в [3] для описания цепи поставки, были изменены для отражения применяемого в настоящее время словаря терминов. поставщик - организация - потребитель Термин «организация» заменяет термин «поставщик», используемый в первой редакции [3], и относится к объекту, в отношении которого применяется настоящий стандарт. Кроме того, термин «поставщик» заменяет термин «субподрядчик». В тексте настоящего стандарта термин «продукция» может означать также «услугу» |
3 Термины и определения В настоящем стандарте применяют термины и определения, данные в ИСО 9000:2000, а также следующие термины с соответствующими определениями. Приведенные ниже термины, используемые в настоящем стандарте для описания цепи поставки, были изменены для отражения применяемого в настоящее время словаря терминов. поставщик - организация - потребитель Термин «организация» заменяет термин «поставщик», используемый в первой редакции [3], и относится к объекту, в отношении которого применяется настоящий стандарт. Кроме того, термин «поставщик» заменяет термин «субподрядчик». В тексте настоящего стандарта термин «продукция» может означать также «услугу». Там, где требования определены как применимые к «медицинским изделиям», эти же требования равнозначно применимы и к связанным с ними услугам, предоставляемым той же организацией. Термины и определения, помещенные ниже, следует рассматривать как наиболее общие, поскольку термины и определения, существующие в национальных нормативных документах, могут отличаться от них и быть предпочтительными. Причины различий: раздел 3 относится к медицинским изделиям, он также включает предупреждение о том, что национальные (региональные) нормативные документы могут содержать определения, заменяющие определения в соответствии с 3.1 - 3.8 или на которые приводится ссылка в ИСО 13485:2003. Определения в соответствии с 3.1 - 3.8 предназначены для медицинских изделий |
|
4 Система менеджмента качества 4.1 Общие требования Организация должна разработать, документировать, внедрить и поддерживать в рабочем состоянии систему менеджмента качества, постоянно улучшать ее результативность в соответствии с требованиями настоящего стандарта. Организация должна: а) определять процессы, необходимые для системы менеджмента качества, и их применение во всей организации (1.2); б) определять последовательность и взаимодействие этих процессов; в) определять критерии и методы, необходимые для обеспечения результативности как при осуществлении, так и при управлении этими процессами; г) обеспечивать наличие ресурсов и информации, необходимых для поддержки этих процессов и их мониторинга; д) осуществлять мониторинг, измерение и анализ этих процессов; е) принимать меры, необходимые для достижения запланированных результатов и постоянного улучшения этих процессов. Организация должна осуществлять менеджмент этих процессов в соответствии с требованиями настоящего стандарта. Если организация решает передать сторонним организациям выполнение какого-либо процесса, влияющего на соответствие продукции требованиям, она должна обеспечивать со своей стороны контроль такого процесса. Управление им должно быть определено в системе менеджмента качества. Примечание - В процессы, необходимые для системы менеджмента качества, следует включать процессы управленческой деятельности руководства, обеспечения ресурсами, процессы жизненного цикла продукции и измерения |
4 Система менеджмента качества 4.1 Общие требования Организация должна разрабатывать, документировать, внедрять и поддерживать в рабочем состоянии систему менеджмента качества, а также поддерживать ее результативность в соответствии с требованиями настоящего стандарта. Организация должна: а) определять процессы, необходимые для системы менеджмента качества, и их применение во всей организации (1.2); б) определять последовательность и взаимодействие этих процессов; в) определять критерии и методы, необходимые для обеспечения результативности как при осуществлении, так и при управлении этими процессами; г) обеспечивать наличие ресурсов и информации, необходимых для поддержки этих процессов и их мониторинга; д) осуществлять мониторинг, измерение и анализ этих процессов; е) выполнять действия, необходимые для достижения запланированных результатов и поддержания результативности этих процессов. Организация должна осуществлять менеджмент этих процессов в соответствии с требованиями настоящего стандарта. Если организация решает передать сторонним организациям выполнение какого-либо процесса, влияющего на соответствие продукции требованиям, она должна обеспечивать со своей стороны контроль такого процесса. Управление им должно быть определено в системе менеджмента качества (8.5.1). Примечание - В процессы, необходимые для системы менеджмента качества, следует включать процессы управленческой деятельности руководства, обеспечения ресурсами, процессы жизненного цикла продукции и измерения. Причина различий: пункт 4.1 отражает требования действующих в мире нормативных документов и облегчает всемирную гармонизацию новых требований, установленных к медицинским изделиям. Требования действующих нормативных документов направлены на применение результативной системы менеджмента качества для обеспечения производства безопасной и эффективно функционирующей продукции |
|
4.2 Требования к документации 4.2.1 Общие положения Документация системы менеджмента качества должна включать: а) документально оформленные заявления о политике и целях в области качества; б) руководство по качеству; в) документированные процедуры, требуемые настоящим стандартом; г) документы, необходимые организации для обеспечения эффективного планирования, осуществления процессов и управления ими; д) записи, требуемые настоящим стандартом (4.2.4) Примечания 1 Там, где в настоящем стандарте встречается термин «документированная процедура», это означает что процедура разработана, документально оформлена внедрена и поддерживается в рабочем состоянии. 2 Степень документированности системы менеджмента качества одной организации может отличаться от другой в зависимости от: а) размера организации и вида деятельности; б) сложности и взаимодействия процессов; в) компетенции персонала. 3 Документация может быть в любой форме и на любом носителе |
4.2 Требования к документации 4.2.1 Общие положения Документация системы менеджмента качества должна включать: а) документально оформленные заявления о политике и целях в области качества; б) руководство по качеству; в) документированные процедуры, требуемые настоящим стандартом; г) документы, необходимые организации для обеспечения эффективного планирования, осуществления процессов и управления ими; д) записи, требуемые настоящим стандартом (4.2.4). е) другие документы, указанные в национальных или региональных нормативных документах. Там, где в настоящем стандарте указано, что требование, процедура, какая-либо деятельность или специальное мероприятие должны быть «документированы», подразумевается, что их также следует выполнять и поддерживать в рабочем состоянии. Для каждого типа или модели медицинского изделия организация должна создать и поддерживать в рабочем состоянии файл, содержащий или идентифицирующий документы, определяющие спецификации на продукцию и требования к системе менеджмента качества (4.2.3). Эти документы должны также определять полный процесс изготовления и, если необходимо, монтажа и обслуживания медицинского изделия. Примечания 1 Степень документированности системы менеджмента качества одной организации может отличаться от другой в зависимости от: а) размера организации и вида деятельности; б) сложности и взаимодействия процессов; в) компетенции персонала. 2 Документация может быть в любой форме и на любом носителе. Причины различий: подпункт 4.2.1 ИСО 13485:2003 включает требования, содержащиеся в соответствующем подпункте [3], дополненные требованиями, установленными к документации, а также специальными требованиями к файлу, содержащему конкретные документы для каждого типа/модели медицинского изделия. Кроме того, подпункт 4.2.1 содержит требования к документации для различных видов деятельности и специальные положения. Этот подпункт отражает положения действующих в мире нормативных документов и облегчает всемирную гармонизацию новых требований, установленных к медицинским изделиям |
|
4.2.2 Руководство по качеству Организация должна разработать и поддерживать в рабочем состоянии руководство по качеству, содержащее: а) область применения системы менеджмента качества, включая подробности и обоснование любых исключений (1.2); б) документированные процедуры, разработанные для системы менеджмента качества, или ссылки на них; в) описание взаимодействия процессов системы менеджмента качества |
4.2.2 Руководство по качеству Организация должна разработать и поддерживать в рабочем состоянии руководство по качеству, содержащее: а) область применения системы менеджмента качества, включая подробности и обоснование любых исключений и/или неприменения (1.2); б) документированные процедуры, разработанные для системы менеджмента качества, или ссылки на них; в) описание взаимодействия процессов системы менеджмента качества. Руководство по качеству должно определять структуру документов, используемых в системе менеджмента качества |
|
4.2.3 Управление документацией Документами системы менеджмента качества необходимо управлять. Записи - специальный вид документов, и ими надо управлять согласно требованиям, приведенным в 4.2.4. Для определения необходимых средств управления должна быть разработана документированная процедура, предусматривающая: а) проверку документов на адекватность до их выпуска; б) анализ и актуализацию, по мере необходимости, и переутверждение документов; в) обеспечение идентификации изменений и статуса пересмотра документов; г) обеспечение наличия соответствующих версий документов в местах их применения; д) обеспечение сохранения документов четкими и легко идентифицируемыми; е) обеспечение идентификации документов внешнего происхождения и управление их рассылкой; ж) предотвращение непреднамеренного использования устаревших документов и применение соответствующей идентификации таких документов, оставленных для каких-либо целей |
4.2.3 Управление документацией Документами системы менеджмента качества необходимо управлять. Записи - специальный вид документов, и ими надо управлять согласно требованиям, приведенным в 4.2.4. Для определения необходимых средств управления должна быть разработана документированная процедура, предусматривающая: а) проверку и утверждение документов на адекватность до их выпуска; б) анализ и актуализацию, по мере необходимости, и переутверждение документов; в) обеспечение идентификации изменений и статуса пересмотра документов; г) обеспечение наличия соответствующих версий документов в местах их применения; д) обеспечение сохранения документов четкими и легко идентифицируемыми; е) обеспечение идентификации документов внешнего происхождения и управление их рассылкой; ж) предотвращение непреднамеренного использования устаревших документов и применение соответствующей идентификации таких документов, оставленных для каких-либо целей. Организация должна обеспечивать анализ и утверждение изменений в документах либо должностным лицом, утверждавшим первоначальный документ, либо другим специально назначенным должностным лицом, имеющим доступ к соответствующей исходной информации, на основании которой принимается решение. Организация должна определять период времени, в течение которого следует хранить не менее одной копии устаревших регулирующих документов. На этот период должен быть обеспечен доступ к документам, в соответствии с которыми медицинское изделие было изготовлено и испытано, в течение, по крайней мере, срока службы изделия, определенного организацией, но не менее срока хранения любой итоговой записи (4.2.4) или согласно установленным требованиям |
|
4.2.4 Управление записями Записи должны вестись и поддерживаться в рабочем состоянии для предоставления свидетельств соответствия требованиям и результативности функционирования системы менеджмента качества. Они должны оставаться четкими, легко идентифицируемыми и восстанавливаемыми. Надо разработать документированную процедуру для определения средств управления, требуемых при идентификации, хранении, защите, восстановлении, определении сроков хранения и изъятии записей |
4.2.4 Управление записями Записи должны вестись и поддерживаться в рабочем состоянии для предоставления свидетельств соответствия требованиям и результативности функционирования системы менеджмента качества. Они должны оставаться четкими, легко идентифицируемыми и восстанавливаемыми. Надо разработать документированную процедуру для определения средств управления, требуемых при идентификации, хранении, защите, восстановлении, определении сроков хранения и изъятии записей. Организация должна хранить записи в течение, по крайней мере, срока службы медицинского изделия, определенного организацией, но не менее двух лет с момента выпуска изделия организацией или в соответствии с установленными требованиями |
|
5 Ответственность руководства 5.1 Обязательства руководства Высшее руководство должно обеспечивать наличие свидетельств принятия обязательств по разработке и внедрению системы менеджмента качества, а также постоянному улучшению ее результативности посредством: а) доведения до сведения организации важности выполнения требований потребителя, а также законодательных и обязательных требований; б) разработки политики в области качества; в) обеспечения разработки целей в области качества; г) проведения анализа со стороны руководства; д) обеспечения необходимыми ресурсами |
5 Ответственность руководства 5.1 Обязательства руководства Высшее руководство организации должно подтверждать свою приверженность разработке и внедрению системы менеджмента качества, а также поддержанию ее результативности посредством: а) доведения до сведения организации важности выполнения требований потребителя, а также законодательных и обязательных требований; б) разработки политики в области качества; в) обеспечения разработки целей в области качества; г) проведения анализа со стороны руководства; д) обеспечения необходимыми ресурсами. Примечание - В настоящем стандарте установленные требования ограничены только безопасностью и функционированием конкретного медицинского изделия. Причины различий: пункт 5.1 отражает требования действующих в мире нормативных документов и облегчает всемирную гармонизацию новых требований, установленных к медицинским изделиям. Действующие нормативные документы направлены на применение результативной системы менеджмента качества для обеспечения производства безопасной и эффективно функционирующей продукции |
|
5.2 Ориентация на потребителя Высшее руководство должно обеспечивать определение и выполнение требований потребителей для повышения их удовлетворенности (7.2.1 и 8.2.1) |
5.2 Ориентация на потребителя Высшее руководство организации должно обеспечивать определение и выполнение требований потребителя (7.2.1 и 8.2.1). Причины различий: изменение текста соответствует такой ситуации, когда выполнение требований потребителя не является целью производства медицинских изделий, в результате чего настоящий пункт соответствует цели настоящего стандарта, которая состоит в облегчении всемирной гармонизации требований, установленных к системам управления качеством |
|
5.3 Политика в области качества Высшее руководство должно обеспечивать, чтобы политика в области качества: а) соответствовала целям организации; б) включала обязательство соответствовать требованиям и постоянно повышать результативность системы менеджмента качества; в) создавала основы для постановки и анализа целей в области качества; г) была доведена до сведения персонала организации и понятна ему; д) анализировалась на постоянную пригодность |
5.3 Политика в области качества Высшее руководство должно обеспечивать, чтобы политика в области качества: а) соответствовала целям организации; б) включала обязательство соответствовать требованиям системы менеджмента качества и поддерживать ее результативность; в) создавала основы для постановки и анализа целей в области качества; г) была доведена до сведения персонала организации и понятна ему; д) анализировалась на постоянную пригодность. Причины различий: из ИСО 13485:2003, пункт 5.3, перечисление б) исключено обязательство постоянного повышения результативности системы менеджмента качества, которое заменено обязательством поддержания результативности системы менеджмента качества. Такая замена соответствует цели действующих в мире нормативных документов и предназначена для облегчения всемирной гармонизации установленных требований к системам менеджмента качества |
|
- |
5.4 Планирование [Пункт 5.4 ИСО 13485:2003 аутентичен пункту 5.4 [3] |
|
5.5 Ответственность, полномочия и обмен информацией 5.5.1 Ответственность и полномочия Высшее руководство должно обеспечивать определение и доведение до сведения персонала организации ответственности и полномочий |
5.5 Ответственность, полномочия и обмен информацией 5.5.1 Ответственность и полномочия Высшее руководство организации должно обеспечивать: определение ответственности и полномочий, которые необходимо документировать, и доведение их до сведения персонала организации; взаимодействие и независимость персонала, руководящего, выполняющего и верифицирующего работу по обеспечению качества, и определять полномочия, необходимые для выполнения этих задач. Особое внимание следует уделить назначению конкретных лиц, ответственных за виды деятельности, связанные с наблюдением за эксплуатацией готовых изделий, а также оформлением отчета об инцидентах (8.2.1 и 8.5.2) |
|
5.5.2 Представитель руководства Высшее руководство должно назначить представителя из состава руководства, который независимо от других обязанностей должен нести ответственность и иметь полномочия, распространяющиеся на: а) обеспечение разработки, внедрения и поддержания в рабочем состоянии процессов, требуемых системой менеджмента качества; б) представление отчетов высшему руководству о функционировании системы менеджмента качества и необходимости улучшения; в) содействие распространению понимания требований потребителей по всей организации. Примечание - В ответственность представителя руководства может быть включено поддержание связи с внешними сторонами по вопросам, касающимся системы менеджмента качества |
5.5.2 Представитель руководства Высшее руководство должно назначить представителя из состава руководства, который независимо от других обязанностей должен нести ответственность и иметь полномочия, распространяющиеся на: а) обеспечение разработки, внедрения и поддержания в рабочем состоянии процессов, требуемых системой менеджмента качества; б) представление отчетов высшему руководству о функционировании системы менеджмента качества и необходимости улучшения (8.5); в) обеспечение внутри организации понимания установленных требований и требований потребителя. Примечание - В ответственность представителя руководства может быть включено поддержание связи с внешними сторонами по вопросам, касающимся системы менеджмента качества |
|
- |
5.5.3 Внутренний обмен информацией [Подпункт 5.5.3 ИСО 13485:2003 аутентичен подпункту 5.5.3 [3] |
|
5.6 Анализ со стороны руководства |
5.6 Анализ со стороны руководства 5.6.1 Общие положения [Подпункт 5.6.1 ИСО 13485:2003 аутентичен подпункту 5.6.1 [3] |
|
5.6.2 Входные данные для анализа Входные данные для анализа со стороны руководства должны включать следующую информацию: а) результаты аудитов (проверок); б) обратную связь с потребителями; в) функционирование процессов и соответствие продукции; г) статус предупреждающих и корректирующих действий; д) последующие действия, вытекающие из предыдущего анализа со стороны руководства; е) изменения, которые могли бы повлиять на систему менеджмента качества; ж) рекомендации по улучшению |
5.6.2 Входные данные для анализа Входные данные для анализа со стороны руководства должны включать следующую информацию: а) результаты аудитов (проверок); б) обратную связь с потребителями; в) функционирование процессов и соответствие продукции; г) статус предупреждающих и корректирующих действий; д) последующие действия, вытекающие из предыдущего анализа со стороны руководства; е) изменения, которые могли бы повлиять на систему менеджмента качества; ж) рекомендации по улучшению; з) новые или пересмотренные установленные требования |
|
5.6.3 Выходные данные анализа Выходные данные анализа со стороны руководства должны включать все решения и действия, относящиеся к: а) повышению результативности системы менеджмента качества и ее процессов; б) улучшению продукции согласно требованиям потребителей; в) потребности в ресурсах |
5.6.3 Выходные данные анализа Выходные данные анализа со стороны руководства должны включать все решения и действия, относящиеся к: а) улучшениям, необходимым для поддержания результативности системы менеджмента качества и ее процессов; б) улучшению продукции согласно требованиям потребителей; в) потребности в ресурсах |
|
6 Менеджмент ресурсов 6.1 Обеспечение ресурсами Организация должна определять и обеспечивать ресурсы, требуемые для: а) внедрения и поддержания в рабочем состоянии системы менеджмента качества, а также постоянного повышения ее результативности; б) повышения удовлетворенности потребителей путем выполнения их требований |
6 Менеджмент ресурсов 6.1 Обеспечение ресурсами Организация должна определять и обеспечивать ресурсы, требуемые для: а) внедрения системы менеджмента качества и поддержания ее результативности; б) удовлетворения установленных требований и требований потребителя |
|
6.2 Человеческие ресурсы |
6.2 Человеческие ресурсы 6.2.1 Общие положения [Подпункт 6.2.1 ИСО 13485:2003 аутентичен подпункту 6.2.1 [3] |
|
6.2.2 Компетентность, осведомленность и подготовка Организация должна: а) определять необходимую компетентность персонала, выполняющего работу, которая влияет на качество продукции; б) обеспечивать подготовку или предпринимать другие действия с целью удовлетворения этих потребностей; в) оценивать результативность предпринятых мер; г) обеспечивать осведомленность своего персонала об актуальности и важности его деятельности и вкладе в достижение целей в области качества; д) поддерживать в рабочем состоянии соответствующие записи об образовании, подготовке, навыках и опыте (4.2.4) |
6.2.2 Компетентность, осведомленность и подготовка Организация должна: а) определять необходимую компетентность персонала, выполняющего работу, которая влияет на качество продукции; б) обеспечивать подготовку или предпринимать другие действия с целью удовлетворения этих потребностей; в) оценивать результативность предпринятых мер; г) обеспечивать осведомленность своего персонала об актуальности и важности его деятельности и вкладе в достижение целей в области качества; д) поддерживать в рабочем состоянии соответствующие записи об образовании, подготовке, навыках и опыте (4.2.4). Примечание - Национальные или региональные нормативные документы могут требовать от конкретной организации разработки документированных процедур для определения необходимости подготовки персонала |
|
6.3 Инфраструктура Организация должна определять, обеспечивать и поддерживать в рабочем состоянии инфраструктуру, необходимую для достижения соответствия требованиям к продукции. Инфраструктура может включать: а) здания, рабочее пространство и связанные с ним средства труда; б) оборудование для процессов (как технические, так и программные средства); с) службы обеспечения (например транспорт или связь) |
6.3 Инфраструктура Организация должна определять, обеспечивать и поддерживать в рабочем состоянии инфраструктуру, необходимую для достижения соответствия требованиям к продукции. Инфраструктура может включать: а) здания, рабочее пространство и связанные с ним средства труда; б) оборудование для процессов (как технические, так и программные средства); с) службы обеспечения (например транспорт или связь). Организация должна разработать документированные требования к действиям, поддерживающим ее инфраструктуру, включая частоту их проведения, если эти действия или их отсутствие могут повлиять на качество продукции. Записи об этих действиях должны поддерживаться в рабочем состоянии (4.2.4) |
|
6.4 Производственная среда Организация должна создавать производственную среду, необходимую для достижения соответствия требованиям к продукции, и управлять ею |
6.4 Производственная среда Организация должна создавать производственную среду, необходимую для достижения соответствия требованиям к продукции, и управлять ею. Это включает следующие требования: а) организация должна разработать документированные требования к состоянию здоровья, гигиене и одежде персонала, если контакт между персоналом и продукцией или производственной средой может отрицательно повлиять на качество продукции (7.5.1.2.1); б) если производственная среда может отрицательно сказаться на качестве продукции, организация должна разработать документированные требования к производственной среде и документированные процедуры или производственные инструкции для мониторинга и контроля производственной среды (7.5.1.2.1); в) организация должна гарантировать соответствующую подготовку персонала, временно работающего в особых условиях производственной среды, или его нахождение под наблюдением подготовленного лица (6.2.2, перечисление б); г) при необходимости разработку и документирование специальных мер для контроля загрязненной или потенциально загрязненной продукции с целью предотвращения загрязнения остальной продукции, производственной среды или загрязнения персонала (7.5.3.1) |
|
7 Процессы жизненного цикла продукции 7.1 Планирование процессов жизненного цикла продукции Организация должна планировать и разрабатывать процессы, необходимые для обеспечения жизненного цикла продукции. Планирование процессов жизненного цикла продукции должно быть согласовано с требованиями к другим процессам системы менеджмента качества (4.1). При планировании процессов жизненного цикла продукции организация должна установить, если это целесообразно: а) цели в области качества и требования к продукции; б) потребность в разработке процессов, документов, а также в обеспечении ресурсами для конкретной продукции; в) необходимую деятельность по верификации и валидации, мониторингу, контролю и испытаниям для конкретной продукции, а также критерии приемки продукции; г) записи, необходимые для обеспечения свидетельства того, что процессы жизненного цикла продукции и произведенная продукция соответствуют требованиям (4.2.4). Результаты этого планирования должны быть в форме, соответствующей практике организации. Примечания 1 Документ, определяющий процессы системы менеджмента качества (включая процессы жизненного цикла продукции) и ресурсы, которые предстоит применять к конкретной продукции, проекту или контракту, может рассматриваться как план качества 2 При разработке процессов жизненного цикла продукции организация может также применять требования 7.3 |
7 Процессы жизненного цикла продукции 7.1 Планирование процессов жизненного цикла продукции Организация должна планировать и разрабатывать процессы, необходимые для обеспечения жизненного цикла продукции. Планирование процессов жизненного цикла продукции должно быть согласовано с требованиями к другим процессам системы менеджмента качества (4.1). При планировании процессов жизненного цикла продукции организация должна установить, если это целесообразно: а) цели в области качества и требования к продукции; б) потребность в разработке процессов, документов, а также в обеспечении ресурсами для конкретной продукции; в) необходимую деятельность по верификации и валидации, мониторингу, контролю и испытаниям для конкретной продукции, а также критерии приемки продукции; г) записи, необходимые для обеспечения свидетельства того, что процессы жизненного цикла продукции и произведенная продукция соответствуют требованиям (4.2.4). Результаты этого планирования должны быть в форме, соответствующей практике организации. Организация должна разработать документированные требования к управлению риском на всех этапах жизненного цикла продукции. Записи по управлению риском должны поддерживаться в рабочем состоянии (4.2.4 и примечание 3). Примечания 1 Документ, определяющий процессы системы менеджмента качества (включая процессы жизненного цикла продукции) и ресурсы, которые предстоит применять к конкретной продукции, проекту или контракту, может рассматриваться как план качества. 2 При разработке процессов жизненного цикла продукции организация может также применять требования 7.3. 3 См. [5] по управлению риском. Причины различий: пункт 7.1 ИСО 13485:2003 отражает требования действующих в мире нормативных документов и облегчает всемирную гармонизацию новых требований, установленных к медицинским изделиям. Управление риском является ключевым видом деятельности, определяющим характер и объем работы во многих областях, связанных с системой менеджмента качества в организациях, производящих медицинские изделия |
|
7.2 Процессы, связанные с потребителями |
7.2 Процессы, связанные с потребителями 7.2.1 Определение требований, относящихся к продукции [Подпункт 7.2.1 ИСО 13485:2003 аутентичен подпункту 7.2.1 [3] |
|
7.2.2 Анализ требований, относящихся к продукции Организация должна анализировать требования, относящиеся к продукции. Этот анализ должен проводиться до принятия организацией обязательства поставлять продукцию потребителю (например участия в тендерах, принятия контрактов или заказов, принятия изменений к контрактам или заказам) и должен обеспечивать: а) определение требований к продукции; б) согласование требований контракта или заказа, отличающихся от ранее сформулированных; в) способность организации удовлетворять определенные требования. Записи результатов анализа и последующих действий, вытекающих из анализа, должны поддерживаться в рабочем состоянии (4.2.4). Если потребители не выдвигают документированных требований, организация должна подтвердить их у потребителя до принятия к исполнению. Если требования к продукции изменены, организация должна обеспечить, чтобы соответствующие документы были исправлены, а заинтересованный персонал был поставлен в известность об изменившихся требованиях. Примечание - В некоторых ситуациях, таких как продажи, осуществляемые через Интернет, практически нецелесообразно проводить официальный анализ каждого заказа. Вместо этого анализ может распространяться на соответствующую информацию о продукции, такую как каталоги или рекламные материалы |
7.2.2 Анализ требований, относящихся к продукции Организация должна анализировать требования, относящиеся к продукции. Этот анализ должен проводиться до принятия организацией обязательства поставлять продукцию потребителю (например участия в тендерах, принятия контрактов или заказов, принятия изменений к контрактам или заказам) и должен обеспечивать: а) определение и документирование требований к продукции; б) согласование требований контракта или заказа, отличающихся от ранее сформулированных; в) способность организации удовлетворять определенные требования. Записи результатов анализа и последующих действий, вытекающих из анализа, должны поддерживаться в рабочем состоянии (4.2.4). Если потребители не выдвигают документированных требований, организация должна подтвердить их у потребителя до принятия к исполнению. Если требования к продукции изменены, организация должна обеспечить, чтобы соответствующие документы были исправлены, а заинтересованный персонал был поставлен в известность об изменившихся требованиях. Примечание - В некоторых ситуациях, таких как продажи, осуществляемые через Интернет, практически нецелесообразно проводить официальный анализ каждого заказа. Вместо этого анализ может распространяться на соответствующую информацию о продукции, такую как каталоги или рекламные материалы |
|
7.2.3 Связь с потребителями Организация должна определять и осуществлять эффективные меры по поддержанию связи с потребителями, касающиеся: а) информации о продукции; б) прохождения запросов, контракта или заказа, включая поправки; в) обратной связи с потребителями, включая жалобы потребителей |
7.2.3 Связь с потребителями Организация должна определять и осуществлять эффективные меры по поддержанию связи с потребителями, касающиеся: а) информации о продукции; б) прохождения запросов, контракта или заказа включая поправки; в) обратной связи с потребителями, включая жалобы потребителей (8.2.1); г) пояснительных уведомлений (8.5.1) |
|
7.3 Проектирование и разработка 7.3.1 Планирование проектирования и разработки Организация должна планировать и управлять проектированием и разработкой продукции. В ходе планирования проектирования и разработки организация должна устанавливать: а) стадии проектирования и разработки; б) проведение анализа, верификацию и валидацию, соответствующие каждой стадии проектирования и разработки; в) ответственность и полномочия в области проектирования и разработки. Организация должна управлять взаимодействием различных групп, занятых проектированием и разработкой, с целью обеспечения эффективной связи и четкого распределения ответственности. Результаты планирования должны актуализироваться, если это целесообразно, по ходу проектирования и разработки (4.2.3); |
7.3 Проектирование и разработка 7.3.1 Планирование проектирования и разработки Организация должна разрабатывать документированные процедуры проектирования и разработки медицинских изделий. Организация обязана планировать и управлять проектированием и разработкой продукции. В ходе планирования проектирования и разработки организация должна устанавливать: а) стадии проектирования и разработки б) виды деятельности по анализу, верификации, валидации и перемещению проекта внутри организации (см. примечание), соответствующие каждой стадии проектирования и разработки; в) ответственность и полномочия в области проектирования и разработки. Организация должна управлять взаимодействием различных групп, занятых проектированием и разработкой, с целью обеспечения эффективной связи и четкого распределения ответственности. Результаты планирования должны быть документированы и, если необходимо, актуализированы при совершенствовании проектирования и разработки (4.2.3) Примечание - Деятельность по передаче проекта в процессе проектирования и разработки медицинского изделия должна гарантировать, что выходные данные проектирования и разработки будут верифицированы как соответствующие требованиям к процессу изготовления, прежде чем станут окончательными спецификациями продукции. Причины различий: подпункт 7.3.1 ИСО 13485:2003 отражает требования действующих в мире нормативных документов и облегчает всемирную гармонизацию новых требований, установленных к медицинским изделиям. В целом, требования ИСО 13485:2003 к документированию процедур соответствуют требованиям [3], а также требованиям большинства национальных (региональных) нормативных документов |
|
7.3.2 Входные данные для проектирования и разработки Входные данные, относящиеся к требованиям к продукции, должны быть определены, а записи должны поддерживаться в рабочем состоянии (4.2.4). Входные данные должны включать: а) функциональные и эксплуатационные требования; б) соответствующие законодательные и другие обязательные требования; в) там, где это целесообразно, информацию, взятую из предыдущих аналогичных проектов; г) другие требования, важные для проектирования и разработки. Входные данные должны анализироваться на достаточность. Требования должны быть полными, недвусмысленными и непротиворечивыми |
7.3.2 Входные данные для проектирования и разработки Входные данные, относящиеся к требованиям к продукции, должны быть определены, а записи должны поддерживаться в рабочем состоянии (4.2.4). Входные данные должны включать: а) функциональные и эксплуатационные требования, требования к безопасности согласно назначению; б) соответствующие законодательные и другие обязательные требования; в) там, где это целесообразно, информацию, взятую из предыдущих аналогичных проектов; г) другие требования, важные для проектирования и разработки; д) выходные данные по управлению риском (7.1). Входные данные должны быть проанализированы на соответствие установленным требованиям и утверждены. Требования должны быть полными, недвусмысленными и непротиворечивыми |
|
7.3.3 Выходные данные проектирования и разработки Выходные данные проектирования и разработки должны быть представлены в форме, позволяющей провести верификацию относительно входных требований к проектированию и разработке, а также должны быть утверждены до их последующего использования. Выходные данные проектирования и разработки должны: а) соответствовать входным требованиям к проектированию и разработке; б) обеспечивать соответствующей информацией по закупкам, производству и обслуживанию; в) содержать критерии приемки продукции или ссылки на них; г) определять характеристики продукции, существенные для ее безопасного и правильного использования |
7.3.3 Выходные данные проектирования и разработки Выходные данные проектирования и разработки должны быть представлены в форме, позволяющей провести верификацию относительно входных требований к проектированию и разработке, а также должны быть утверждены до их последующего использования. Выходные данные проектирования и разработки должны: а) соответствовать входным требованиям к проектированию и разработке; б) обеспечивать соответствующей информацией по закупкам, производству и обслуживанию; в) содержать критерии приемки продукции или ссылки на них; г) определять характеристики продукции, существенные для ее безопасного и правильного использования Записи выходных данных проектирования и разработки должны поддерживаться в рабочем состоянии (4.2.4) Примечание - Записи выходных данных проектирования и разработки могут включать спецификации, производственные процессы, технические чертежи и журналы технических записей или исследований |
|
7.3.4 Анализ проекта и разработки На тех стадиях, где это целесообразно, должен проводиться систематический анализ проекта и разработки в соответствии с запланированными мероприятиями (7.3.1) с целью: а) оценивания способности результатов проектирования и разработки удовлетворять требованиям; б) выявления любых проблем и внесения предложений по необходимым действиям. В состав участников такого анализа должны включаться представители подразделений, имеющих отношение к анализируемой (ым) стадии (ям) проектирования и разработки. Записи результатов анализа и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4) |
7.3.4 Анализ проекта и разработки На тех стадиях, где это целесообразно, должен проводиться систематический анализ проекта и разработки в соответствии с запланированными мероприятиями (7.3.1) с целью: а) оценки способности результатов проектирования и разработки удовлетворять требованиям; б) выявления любых проблем и внесения предложений по необходимым действиям. Такой анализ должны выполнять представители служб, имеющих отношение к анализируемым стадиям проекта и разработки, а также другие специалисты организации (5.5.1 и 6.2.1). Записи результатов анализа и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4) |
|
- |
7.3.5 Верификация проекта и разработки [Подпункт 7.3.5 ИСО 13485:2003 аутентичен подпункту 7.3.5 [3] |
|
7.3.6 Валидация проекта и разработки Валидация проекта и разработки должна осуществляться в соответствии с запланированными мероприятиями (7.3.1), чтобы удостовериться, что полученная в результате продукция соответствует требованиям к установленному или предполагаемому использованию, если оно известно. Где это практически целесообразно, валидация должна быть завершена до поставки или применения продукции. Записи результатов валидации и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4) |
7.3.6 Валидация проекта и разработки Валидация проекта и разработки должна проводиться в соответствии с запланированными мероприятиями (7.3.1) для обеспечения соответствия готовой продукции требованиям к ее назначению или специальному применению. Валидация должна быть проведена до поставки продукции или введения ее в эксплуатацию (см. примечание 1). Записи результатов валидации и всех необходимых действий должны поддерживаться в рабочем состоянии (4.2.4) Организация обязана проводить клинические и/или эксплуатационные испытания медицинских изделий как часть мероприятий по валидации проекта и разработки в соответствии с требованиями национальных или региональных нормативных документов (см. примечание 2). Примечания 1 Если медицинское изделие подлежит валидации только после сборки и монтажа на месте его применения, поставка продукции не считается законченной до тех пор, пока продукция не будет формально передана потребителю. 2 Представление медицинского изделия для проведения клинических и/или эксплуатационных испытаний не считается поставкой |
|
- |
7.3.7 Управление изменениями проекта и разработки [Подпункт 7.3.7 ИСО 13485:2003 аутентичен подпункту 7.3.7 [3] |
|
7.4 Закупки 7.4.1 Процесс закупок Организация должна обеспечивать соответствие закупленной продукции установленным требованиям к закупкам. Тип и степень управления, применяемые по отношению к поставщику и закупленной продукции, должны зависеть от ее воздействия на последующие стадии жизненного цикла продукции или готовую продукцию. Организация должна оценивать и выбирать поставщиков на основе их способности поставлять продукцию в соответствии с требованиями организации. Должны быть разработаны критерии отбора, оценки и повторной оценки. Записи результатов оценивания и любых необходимых действий, вытекающих из оценки, должны поддерживаться в рабочем состоянии (4.2.4) |
7.4 Закупки 7.4.1 Процесс закупок Организация должна разработать документированные процедуры, обеспечивающие соответствие закупленной продукции установленным требованиям к закупкам. Тип и степень управления, применяемые по отношению к поставщику и закупленной продукции, должны зависеть от ее воздействия на последующие стадии жизненного цикла продукции или готовую продукцию. Организация должна оценивать и выбирать поставщиков на основе их способности поставлять продукцию в соответствии с требованиями организации. Должны быть разработаны критерии отбора, оценки и повторной оценки. Записи результатов оценивания и любых необходимых действий, вытекающих из оценки, должны поддерживаться в рабочем состоянии (4.2.4) |
|
7.4.2 Информация по закупкам Информация по закупкам должна описывать заказанную продукцию, включая, где это необходимо: а) требования к утверждению продукции, процедур, процессов и оборудования; б) требования к квалификации персонала; в) требования к системе менеджмента качества. Организация должна обеспечивать адекватность установленных требований к закупкам до их сообщения поставщику |
7.4.2 Информация по закупкам Информация по закупкам должна описывать заказанную продукцию, включая, где это необходимо: а) требования к утверждению продукции, процедур, процессов и оборудования; б) требования к квалификации персонала; в) требования к системе менеджмента качества. Организация должна обеспечивать адекватность установленных требований к закупкам до их сообщения поставщику. В той степени, в какой это необходимо для осуществления прослеживаемости (7.5.3.2), организация должна поддерживать в рабочем состоянии соответствующую информацию по закупкам, то есть документы (4.2.3) и записи (4.2.4) |
|
7.4.3 Верификация закупленной продукции Организация должна разработать и осуществлять контроль или другую деятельность, необходимую для обеспечения соответствия закупленной продукции установленным требованиям к закупкам. Если организация или ее потребитель предполагают осуществить верификацию на предприятии поставщика, то организация должна установить в информации по закупкам предполагаемые меры по проверке и порядок выпуска продукции у поставщика |
7.4.3 Верификация закупленной продукции Организация должна разработать и осуществлять контроль или другую деятельность, необходимую для обеспечения соответствия закупленной продукции установленным требованиям к закупкам. Если организация или потребитель ее продукции предлагают осуществить верификацию на предприятии поставщика, то организация должна установить в информации по закупкам предполагаемые меры по проверке и порядок выпуска продукции у поставщика Записи по верификации должны поддерживаться в рабочем состоянии (4.2.4) |
|
7.5 Производство и обслуживание 7.5.1 Управление производством и обслуживанием Организация должна планировать и обеспечивать производство и обслуживание в управляемых условиях. Управляемые условия должны включать, если это целесообразно: а) наличие информации, описывающей характеристики продукции; б) наличие рабочих инструкций, в случае необходимости; в) применение подходящего оборудования; г) наличие и применение контрольных и измерительных приборов; д) проведение мониторинга и измерений; е) осуществление выпуска, поставки и действий после поставки продукции |
7.5 Производство и обслуживание 7.5.1 Управление производством и обслуживанием 7.5.1.1 Общие требования Организация должна планировать и обеспечивать производство и обслуживание в управляемых условиях. Управляемые условия должны включать, если это целесообразно: а) наличие информации, описывающей характеристики продукции; б) наличие документированных процедур, рабочих инструкций, эталонных процедур измерения и, если необходимо, справочного материала; в) применение подходящего оборудования; г) наличие и применение контрольных и измерительных приборов; д) проведение мониторинга и измерений; е) осуществление выпуска, поставки и действий после поставки продукции; ж) выполнение конкретных действий по маркировке и упаковке. Организация должна разработать форму записей и поддерживать их в рабочем состоянии (4.2.4) для каждой партии медицинских изделий, чтобы обеспечить их прослеживаемость в соответствии с 7.5.3 и установить количество произведенной продукции и продукции, предназначенной для продажи. Записи по каждой партии изделий должны быть верифицированы и утверждены. Примечание - Партией может считаться одно медицинское изделие. 7.5.1.2 Управление производством и обслуживанием. Специальные требования 7.5.1.2.1 Чистота продукции и контроль загрязненности Организация должна разработать документированные требования к чистоте продукции, если: а) перед стерилизацией и/или применением продукция проходит очистку в организации; б) продукция поставляется в нестерильном виде и подлежит очистке перед стерилизацией и/или применением; в) продукция поставляется в нестерильном виде и ее чистота не имеет значения для применения; г) реагенты для очистки продукции должны быть удалены при ее изготовлении. Если продукция подвергается очистке в соответствии с перечислениями а) или б), требования, содержащиеся в (6.4, перечисление а) и (6.4, перечисление б), перед процедурой очистки не применяются. 7.5.1.2.2 Работы по монтажу При необходимости организация должна разработать документированные требования, содержащие критерии приемки и верификации монтажа медицинского изделия. Если согласованные с потребителем требования позволяют выполнять монтаж не только силами организации или ее полномочного представителя, организация должна разработать документированные требования к такому монтажу и его верификации. Записи по монтажу и верификации, которые осуществляет организация или ее полномочный представитель, должны поддерживаться в рабочем состоянии (4.2.4). 7.5.1.2.3 Деятельность по обслуживанию Если требование к обслуживанию является специальным, организация должна, при необходимости, разработать документированные процедуры, рабочие инструкции, справочные материалы и эталонные процедуры измерения для осуществления обслуживания и его верификации в соответствии со специальным требованием. Записи по обслуживанию, выполняемому организацией, должны поддерживаться в рабочем состоянии (4.2.4). Примечание - Обслуживание может включать, например, ремонт и техническое обслуживание. 7.5.1.3 Специальные требования к стерильным медицинским изделиям Организация должна поддерживать записи по параметрам процессов стерилизации, применяемых для каждой партии стерилизуемой продукции (4.2.4). Записи по стерилизации должны прослеживаться для каждой партии произведенных медицинских изделий (7.5.1.1) |
|
7.5.2 Валидация процессов производства и обслуживания Организация должна подтверждать все процессы производства и обслуживания, результаты которых нельзя проверить посредством последовательного мониторинга или измерения. К ним относятся все процессы, недостатки которых становятся очевидными только после начала использования продукции или после предоставления услуги. Валидация должна продемонстрировать способность этих процессов достигать запланированных результатов. Организация должна разработать меры по этим процессам, включая, если это приемлемо: а) определенные критерии для анализа и утверждения процессов; б) утверждение соответствующего оборудования и квалификации персонала; в) применение конкретных методов и процедур; г) требования к записям (4.2.4); д) повторную валидацию |
7.5.2 Валидация процессов производства и обслуживания 7.5.2.1 Общие требования Организация должна подтверждать все процессы производства и обслуживания, результаты которых нельзя проверить посредством последовательного мониторинга или измерения. К ним относятся все процессы, недостатки которых становятся очевидными только после начала использования продукции. Валидация должна продемонстрировать способность этих процессов достигать запланированных результатов. Организация должна разработать меры по этим процессам, включая, если это приемлемо: а) определенные критерии для анализа и утверждения процессов; б) утверждение соответствующего оборудования и квалификации персонала; в) применение конкретных методов и процедур; г) требования к записям (4.2.4); д) повторную валидацию. Организация должна разрабатывать документированные процедуры валидации применения компьютерного программного обеспечения (и изменений такого обеспечения и/или его применения) при производстве и обслуживании, которые могут оказывать влияние на способность продукции удовлетворять установленным требованиям. Такое программное обеспечение следует утверждать до его первого применения. Записи о результатах валидации должны поддерживаться в рабочем состоянии (4.2.4). 7.5.2.2 Специальные требования к стерильным медицинским изделиям Организация должна разрабатывать документированные процедуры валидации процессов стерилизации. Процессы стерилизации следует утверждать до их первого применения. Записи о результатах валидации процессов стерилизации должны поддерживаться в рабочем состоянии (4.2.4) |
|
7.5.3 Идентификация и прослеживаемость Если это целесообразно, организация должна идентифицировать продукцию при помощи соответствующих средств на всех стадиях ее жизненного цикла. Организация должна идентифицировать статус продукции по отношению к требованиям мониторинга и измерений. Если прослеживаемость является требованием, то организация должна управлять специальной идентификацией продукции и регистрировать ее (4.2.4). Примечание - В ряде отраслей промышленности менеджмент конфигурации является средством, с помощью которого поддерживается идентификация и прослеживаемость |
7.5.3 Идентификация и прослеживаемость 7.5.3.1 Идентификация Организация должна идентифицировать продукцию на протяжении всего жизненного цикла с помощью соответствующих средств и разрабатывать документированные процедуры для такой идентификации продукции. Организация должна разрабатывать документированные процедуры, гарантирующие, что медицинские изделия, возвращенные в организацию как несоответствующие, идентифицированы и отделены от изделий, соответствующих установленным требованиям (6.4, перечисление г). 7.5.3.2 Прослеживаемость 7.5.3.2.1 Общие положения Организация должна разрабатывать документированные процедуры прослеживаемости. Такие процедуры должны определять степень прослеживаемости продукции и необходимые записи (4.2.4, 8.3 и 8.5). Если прослеживаемость является требованием, то организация должна управлять специальной идентификацией продукции и регистрировать ее (4.2.4). Примечание - Управление конфигурацией продукции является средством, с помощью которого можно осуществлять идентификацию и прослеживаемость 7.5.3.2.2 Специальные требования к активным имплантируемым медицинским изделиям и имплантируемым медицинским изделиям В записи, необходимые для осуществления прослеживаемости, организация должна включать записи обо всех компонентах, материалах и условиях окружающей среды, если они могут явиться причиной того, что медицинское изделие не удовлетворяет установленным к нему требованиям. Организация должна требовать от своих представителей или дистрибьюторов поддерживать записи о распределении медицинских изделий для достижения прослеживаемости и доступности этих записей для контроля. Организация должна обеспечивать регистрацию грузополучателя (наименования и адреса) (4.2.4). 7.5.3.3 Идентификация статуса Организация должна идентифицировать статус продукции по отношению к требованиям мониторинга и измерений. Идентификация статуса продукции должна поддерживаться на всех этапах ее производства, хранения, монтажа и обслуживания для обеспечения отправления, применения или монтажа только продукции, прошедшей все необходимые виды контроля и испытаний (или имеющей официальное разрешение на отклонение от установленных требований) |
|
7.5.4 Собственность потребителей Организация должна проявлять заботу о собственности потребителя, пока она находится под управлением организации или используется ею. Организация должна идентифицировать, верифицировать, защищать и сохранять собственность потребителя, предоставленную для использования или включения в продукцию. Если собственность потребителя утеряна, повреждена или признана непригодной для использования, потребитель должен быть об этом извещен, а записи должны поддерживаться в рабочем состоянии (4.2.4). Примечание - Собственность потребителя может включать в себя интеллектуальную собственность |
7.5.4 Собственность потребителей Организация должна проявлять заботу о собственности потребителя, пока она находится под управлением организации или используется ею. Организация должна идентифицировать, верифицировать, защищать и сохранять собственность потребителя, предоставленную для использования или включения в продукцию. Если собственность потребителя утеряна, повреждена или признана непригодной для использования, потребитель должен быть об этом извещен, а записи должны поддерживаться в рабочем состоянии (4.2.4). Примечание - Собственностью потребителя может быть интеллектуальная собственность или конфиденциальная информация о состоянии его здоровья. |
|
7.5.5 Сохранение соответствия продукции Организация должна сохранять соответствие продукции в ходе внутренней обработки и в процессе поставки к месту назначения. Это сохранение должно включать в себя идентификацию, погрузочно-разгрузочные работы, упаковку, хранение и защиту. Сохранение должно также применяться и к составным частям продукции |
7.5.5 Сохранение соответствия продукции Организация должна разрабатывать документированные процедуры или рабочие инструкции по сохранению соответствия продукции установленным требованиям при осуществлении технологических процессов внутри организации и доставке продукции к месту назначения. Сохранение соответствия должно включать в себя идентификацию, погрузочно-разгрузочные работы, упаковку, хранение и защиту. Сохранение должно также применяться и к составным частям продукции. Организация должна разработать документированные процедуры или рабочие инструкции по управлению продукцией с ограниченным сроком хранения либо продукцией, требующей специальных условий хранения. Такие специальные условия хранения должны регистрироваться и быть управляемыми |
|
7.6 Управление устройствами для мониторинга и измерений Организация должна определить мониторинг и измерения, которые предстоит осуществлять, а также устройства для мониторинга и измерения, необходимые для обеспечения свидетельства соответствия продукции установленным требованиям (7.2.1). Организация должна разработать процессы для подтверждения того, что способ мониторинга и измерения совместим с требованиями к мониторингу и измерениям. Там, где необходимо обеспечивать имеющие законную силу результаты, измерительное оборудование должно быть: а) откалибровано и поверено в установленные периоды или перед его применением по образцовым эталонам, передающим размеры единиц в сравнении с международными или национальными эталонами. При отсутствии таких эталонов база, использованная для калибровки или поверки, должна быть зарегистрирована; б) отрегулировано или повторно отрегулировано по мере необходимости; в) идентифицировано с целью установления статуса калибровки; г) защищено от регулировок, которые сделали бы недействительными результаты измерения; д) защищено от повреждения и ухудшения состояния в ходе обращения, технического обслуживания и хранения. Кроме того, организация должна оценить и зарегистрировать правомочность предыдущих результатов измерения, если обнаружено, что оборудование не соответствует требованиям. Организация должна предпринять соответствующее действие в отношении такого оборудования и любой измеренной продукции. Записи результатов калибровки и поверки должны поддерживаться в рабочем состоянии (4.2.4). Если при мониторинге и измерении установленных требований используют компьютерные программные средства, их способность удовлетворять предполагаемому применению должна быть подтверждена. Это должно быть осуществлено до начала применения и повторно подтверждено по мере необходимости (7.5.2). Примечание - См. [6] для руководства к действию |
7.6 Управление устройствами для мониторинга и измерений Организация должна определить мониторинг и измерения, которые предстоит осуществлять, а также устройства для мониторинга и измерения, необходимые для обеспечения свидетельства соответствия продукции установленным требованиям (7.2.1). Организация должна разработать документированные процедуры обеспечения проведения мониторинга и измерений в соответствии с установленными требованиями. Там, где необходимо обеспечивать имеющие законную силу результаты, измерительное оборудование должно быть: а) откалибровано и поверено в установленные периоды или перед его применением по образцовым эталонам, передающим размеры единиц в сравнении с международными или национальными эталонами. При отсутствии таких эталонов база, использованная для калибровки или поверки, должна быть зарегистрирована; б) отрегулировано или повторно отрегулировано по мере необходимости; в) идентифицировано с целью установления статуса калибровки; г) защищено от регулировок, которые сделали бы недействительными результаты измерения; д) защищено от повреждения и ухудшения состояния в ходе обращения, технического обслуживания и хранения. Кроме того, организация должна оценить и зарегистрировать правомочность предыдущих результатов измерения, если обнаружено, что оборудование не соответствует требованиям. Организация должна предпринять соответствующее действие в отношении такого оборудования и любой измеренной продукции. Записи результатов калибровки и поверки должны поддерживаться в рабочем состоянии (4.2.4). Если при мониторинге и измерении установленных требований используют компьютерные программные средства, их способность удовлетворять предполагаемому применению должна быть подтверждена. Это должно быть осуществлено до начала применения и повторно подтверждено по мере необходимости Примечание - См. руководящие указания в [6] |
|
8 Измерение, анализ и улучшение 8.1 Общие положения Организация должна планировать и применять процессы мониторинга, измерения, анализа и улучшения, необходимые для: а) демонстрации соответствия продукции; б) обеспечения соответствия системы менеджмента качества; с) постоянного повышения результативности системы менеджмента качества. Это должно включать определение применимых методов, в том числе статистических, и область их использования |
8 Измерение (оценка), анализ и улучшение 8.1 Общие положения Организация должна планировать и применять процессы мониторинга, измерения (оценки), анализа и улучшения, необходимые для: а) демонстрации соответствия продукции; б) обеспечения соответствия системы менеджмента качества; с) поддержания результативности системы менеджмента качества. Это должно включать определение применяемых методов, в том числе статистических, и область их использования. Примечание - Национальные или региональные нормативные документы могут содержать требования к разработке документированных процедур для внедрения статистических методов и управления их применением. Причины различий: пункт 8.1 ИСО 13485:2003 отражает требования действующих в мире нормативных документов и облегчает всемирную гармонизацию новых требований, установленных к медицинским изделиям. Действующие нормативные документы направлены на то, чтобы применение результативной системы менеджмента качества обеспечивало производство безопасной и эффективно функционирующей продукции, а не на постоянное совершенствование самой системы менеджмента качества |
|
8.2 Мониторинг и измерение 8.2.1 Удовлетворенность потребителей Организация должна проводить мониторинг информации, касающейся восприятия потребителями соответствия организации требованиям потребителей, как одного из способов измерения работы системы менеджмента качества. Должны быть установлены методы получения и использования этой информации |
8.2 Мониторинг и измерение (оценка) 8.2.1 Обратная связь В качестве одного из способов оценки функционирования системы менеджмента качества организация должна проводить мониторинг информации, относящейся к удовлетворению организацией требований потребителя. Должны быть установлены методы получения и использования этой информации. Организация должна разработать документированную процедуру по системе обратной связи (7.2.3, перечисление с) с целью получения раннего предупреждения о проблемах в области качества и введения в процессы корректирующих и предупреждающих действий (8.5.2 и 8.5.3). Если национальные или региональные нормативные документы требуют от организации накопления опыта на основании информации, полученной после продажи медицинского изделия, анализ такого опыта может стать частью системы обратной связи (8.5.1). Причина различий: понятия «удовлетворенность потребителей» и «восприятие потребителями» слишком субъективны для введения их в нормативные документы в качестве установленного требования. Подпункт 8.2.1 ИСО 13485:2003 отражает требования действующих в мире нормативных документов и облегчает всемирную гармонизацию новых требований, установленных к медицинским изделиям |
|
- |
8.2.2 Внутренние аудиты (проверки) Примечание - См. [7] - руководство по аудиту. [Подпункт 8.2.2 ИСО 13485:2003 аутентичен подпункту 8.2.2 [3], за исключением вышеупомянутого примечания] |
|
- |
8.2.3 Мониторинг и измерение (оценка) процессов [Подпункт 8.2.3 ИСО 13485:2003 аутентичен подпункту 8.2.3 [3] |
|
8.2.4 Мониторинг и измерение продукции Организация должна осуществлять мониторинг и измерять характеристики продукции с целью проверки соблюдения требований к продукции. Это должно осуществляться на соответствующих стадиях процесса жизненного цикла продукции согласно запланированным мероприятиям (7.1). Свидетельства соответствия критериям приемки должны поддерживаться в рабочем состоянии. Записи должны указывать лицо (а), санкционировавшее (ие) выпуск продукции (4.2.4). До завершения всех запланированных мероприятий (7.1) выпуск продукции и предоставление услуги не должны осуществляться, если иное не утверждено соответствующим уполномоченным или, где это применимо, потребителем |
8.2.4 Мониторинг и измерение (оценка) продукции 8.2.4.1 Общие требования Организация должна проводить мониторинг и измерение характеристик продукции с целью верификации соответствия установленным требованиям. Мониторинг и измерение характеристик продукции должны быть проведены на соответствующих стадиях процессов жизненного цикла продукции и в соответствии с запланированными мероприятиями (7.1) и документированными процедурами (7.5.1.1). Свидетельства соответствия критериям приемки должны поддерживаться в рабочем состоянии. Запись должны указывать лицо (а), санкционировавшее (ие) выпуск продукции (4.2.4). Выпуск продукции и обслуживание не могут осуществляться до успешного завершения запланированных мероприятий (7.1). 8.2.4.2 Специальное требование для активных имплантируемых медицинских изделий и имплантируемых медицинских изделий Организация должна вести записи по идентификации персонала (4.2.4), проводящего любые виды контроля или испытаний |
|
8.3 Управление несоответствующей продукцией Организация должна обеспечивать, чтобы продукция, которая не соответствует требованиям, была идентифицирована и управлялась с целью предотвращения непреднамеренного использования или поставки. Средства управления, соответствующая ответственность и полномочия для работы с несоответствующей продукцией должны быть определены в документированной процедуре. Организация должна решать вопрос с несоответствующей продукцией одним или несколькими следующими способами: а) осуществлять действия с целью устранения обнаруженного несоответствия; б) санкционировать ее использование, выпуск или приемку, если имеется разрешение на отклонение от соответствующего полномочного органа или потребителя, где это применимо; в) осуществлять действия с целью предотвращения ее первоначального предполагаемого использования или применения. Записи о характере несоответствий и любых последующих предпринятых действиях, включая полученные разрешения на отклонения, должны поддерживаться в рабочем состоянии (4.2.4). Когда несоответствующая продукция исправлена, она должна быть подвергнута повторной верификации для подтверждения соответствия требованиям. Если несоответствующая продукция выявлена после поставки или начала использования, организация должна предпринять действия, адекватные последствиям (или потенциальным последствиям) несоответствия |
8.3 Управление несоответствующей продукцией Организация должна обеспечивать, чтобы продукция, которая не соответствует требованиям, была идентифицирована и управлялась с целью предотвращения непреднамеренного использования или поставки. Средства управления, соответствующая ответственность и полномочия для работы с несоответствующей продукцией должны быть определены в документированной процедуре. Организация должна решать вопрос с несоответствующей продукцией одним или несколькими следующими способами: а) осуществлять действия с целью устранения обнаруженного несоответствия; б) санкционировать применение, выпуск или приемку продукции при получении разрешения на отклонение от установленных требований; в) осуществлять действия с целью предотвращения ее первоначального предполагаемого использования или применения. Организация должна гарантировать, что несоответствующая продукция будет принята при получении разрешения на отклонение от установленных требований только в том случае, если это допускается установленными требованиями. Записи об идентификации лица (лиц), имеющего (их) полномочия на выдачу разрешений на отклонение, должны поддерживаться в рабочем состоянии (4.2.4). Записи о характере несоответствий и любых последующих предпринятых действиях, включая приемку продукции под разрешение, должны поддерживаться в рабочем состоянии (4.2.4). Когда несоответствующая продукция исправлена, она должна быть подвергнута повторной верификации для подтверждения соответствия требованиям. Если несоответствующая продукция выявлена после поставки или начала использования, организация должна предпринять действия, адекватные последствиям (или потенциальным последствиям) несоответствия. Если продукция нуждается в переработке (один или более раз), организация обязана документировать процесс переработки в рабочей инструкции, на которую следует получить официальное разрешение и одобрение так же, как и на первоначальную рабочую инструкцию. Перед получением официального разрешения и одобрения должны быть определены и документированы (4.2.3 и 7.5.1) любые неблагоприятные последствия переработки продукции |
|
8.4 Анализ данных Организация должна определить, собирать и анализировать соответствующие данные для демонстрации пригодности и результативности системы менеджмента качества, а также оценивания, в какой области можно осуществлять постоянное повышение результативности системы менеджмента качества. Данные должны включать информацию, полученную в результате мониторинга и измерения также и из других соответствующих источников. Анализ данных должен предоставлять информацию по: а) удовлетворенности потребителей (8.2.1); б) соответствию требованиям к продукции (7.2.1); в) характеристикам и тенденциям процессов и продукции, включая возможности проведения предупреждающих действий; г) поставщикам |
8.4 Анализ данных Организация должна разработать документированную процедуру определения, сбора и анализа данных, необходимых для демонстрации результативности системы менеджмента качества и ее соответствия установленным требованиям, а также для оценки возможности повышения ее результативности. Данные должны включать информацию, полученную в результате мониторинга и измерения также и из других соответствующих источников. Анализ данных должен предоставлять информацию по: а) обратной связи (8.2.1); б) соответствию требованиям к продукции (7.2.1); в) характеристикам и тенденциям процессов и продукции, включая возможности проведения предупреждающих действий; г) поставщикам. Записи результатов анализа данных должны поддерживаться в рабочем состоянии (4.2.4) |
|
8.5 Улучшение 8.5.1 Постоянное улучшение Организация должна постоянно повышать результативность системы менеджмента качества посредством использования политики и целей в области качества, результатов аудитов, анализа данных, корректирующих и предупреждающих действий, а также анализа со стороны руководства |
8.5 Улучшение 8.5.1 Общие положения Организация должна идентифицировать и выполнять любые изменения, необходимые для обеспечения и поддержания постоянного соответствия установленным требованиям и результативности системы менеджмента качества, используя политику в области качества, устанавливая цели в этой области, а также используя результаты аудитов, анализа данных, корректирующих и предупреждающих действий и анализа со стороны руководства. Организация должна установить документированные процедуры выпуска и применения пояснительных уведомлений. Следует обеспечить возможность проведения таких процедур в любое время. Записи об изучении жалоб потребителей должны поддерживаться в рабочем состоянии (4.2.4). Если в ходе такого изучения будет установлено, что появлению жалоб потребителей способствует внешняя деятельность организации, между заинтересованными организациями следует провести обмен соответствующей информацией (4.1). Если по любой жалобе потребителя не предприняты корректирующие и/или предупреждающие действия, причину следует объяснить (5.5.1) и зарегистрировать (4.2.4). Если этого требуют национальные или региональные нормативные документы, организация должна разработать документированные процедуры уведомления регулирующих органов о тех инцидентах, которые соответствуют критериям к отчетности. Причина различий: подпункт 8.5.1 ИСО 13485:2003 отражает требования действующих в мире нормативных документов и облегчает всемирную гармонизацию новых требований, установленных к медицинским изделиям. Постоянное совершенствование системы менеджмента качества не является целью действующих нормативных документов |
|
8.5.2 Корректирующие действия Организация должна предпринимать корректирующие действия с целью устранения причин несоответствий для предупреждения повторного их возникновения. Корректирующие действия должны быть адекватными последствиям выявленных несоответствий. Должна быть разработана документированная процедура для определения требований к: а) анализу несоответствий (включая жалобы потребителей); б) установлению причин несоответствий; в) оцениванию необходимости действий, чтобы избежать повторения несоответствий; г) определению и осуществлению необходимых действий; д) записям результатов предпринятых действий (4.2.4); е) анализу предпринятых корректирующих действий |
8.5.2 Корректирующие действия Организация должна предпринимать корректирующие действия с целью устранения причин несоответствий для предупреждения повторного их возникновения. Корректирующие действия должны быть адекватными последствиям выявленных несоответствий. Должна быть разработана документированная процедура для определения требований к: а) анализу несоответствий (включая жалобы потребителей); б) установлению причин несоответствий; в) оцениванию необходимости действий, чтобы избежать повторения несоответствий; г) определению и выполнению необходимых действий, включая, если целесообразно, обновление документации (4.2); д) регистрации результатов любых расследований и принятых мер (4.2.4); е) анализу предпринятых корректирующих действий и их результативности |
|
8.5.3 Предупреждающие действия Организация должна определить действия с целью устранения причин потенциальных несоответствий для предупреждения их появления. Предупреждающие действия должны соответствовать возможным последствиям потенциальных проблем. Должна быть разработана документированная процедура для определения требований к: а) установлению потенциальных несоответствий и их причин; б) оцениванию необходимости действий с целью предупреждения появления несоответствий; в) определению и осуществлению необходимых действий; г) записям результатов предпринятых действий (4.2.4); д) анализу предпринятых предупреждающих действий |
8.5.3 Предупреждающие действия Организация должна определить действия с целью устранения причин потенциальных несоответствий для предупреждения их появления. Предупреждающие действия должны соответствовать возможным последствиям потенциальных проблем. Должна быть разработана документированная процедура для определения требований к: а) установлению потенциальных несоответствий и их причин; б) оцениванию необходимости действий с целью предупреждения появления несоответствий; в) определению и осуществлению необходимых действий; г) регистрации результатов любых расследований и принятых мер (4.2.4); д) анализу предпринятых предупреждающих действий и их результативности |
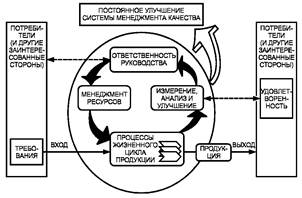
Приложение С
(обязательное)
Сведения о соответствии национальных стандартов ссылочным международным стандартам
Таблица Д.1
|
Обозначение ссылочного международного стандарта |
Обозначение и наименование соответствующего государственного стандарта |
|
ИСО 9000:2000 |
ГОСТ Р ИСО 9000-2001 Системы менеджмента качества. Основные положения и словарь |
|
ИСО 9001:2000 |
ГОСТ Р ИСО 9001-2001 Системы менеджмента качества. Требования |
|
ИСО 9004:2000 |
ГОСТ Р ИСО 9004-2001 Системы менеджмента качества. Рекомендации по улучшению деятельности |
|
ИСО 10012-1:1992 |
* |
|
ИСО 10012-2:1997 |
* |
|
ИСО 13485:1996 |
ГОСТ Р 51536-99 Системы качества. Изделия медицинские. Специальные требования по применению ГОСТ Р ИСО 9001-96 |
|
ИСО 13488:1996 |
ГОСТ Р 51537-99 Системы качества. Изделия медицинские. Специальные требования по применению ГОСТ Р ИСО 9002-96 |
|
ИСО 14001:1996 |
ГОСТ Р ИСО 14001-98 Системы управления окружающей средой. Требования и руководство по применению |
|
ИСО/ТО 14969:1999 |
ГОСТ Р 51538-99 Системы качества. Изделия медицинские. Руководство по применению ГОСТ Р 51536-99 и ГОСТ Р 51537-99 |
|
ИСО 14971:2000 |
ГОСТ Р ИСО 14971.1-99 Медицинские изделия. Управление риском. Часть 1. Применение анализа риска к медицинским изделиям |
|
ИСО 19011:2002 |
ГОСТ Р ИСО 19011-2003 Руководящие указания по аудиту систем менеджмента качества и/или систем экологического менеджмента |
|
* Соответствующий государственный стандарт отсутствует. До его принятия рекомендуется использовать перевод на русский язык данного международного стандарта или гармонизированный с ним национальный (государственный) стандарт страны, на территории которой применяется настоящий стандарт. Информация о наличии перевода данного международного стандарта в национальном фонде стандартов или в ином месте, а также информация о действии на территории страны соответствующего национального (государственного) стандарта может быть приведена в национальных информационных данных, дополняющих настоящий стандарт. |
|
Библиография
[1] ИСО 13485:1996 Системы качества. Изделия медицинские. Специальные требования по применению ИСО 9001:1994
(ISO 13485:1996) (Quality systems. Medical devices. Particular requirements for the application of ISO 9001:1994)
[2] ИСО 13488:1996 Системы качества. Изделия медицинские. Специальные требования по применению ИСО 9002:1996
(ISO 13488:1996) (Quality systems. Medical devices. Particular requirements for the application of ISO 9002:1996)
[3] ИСО 9001:2000 Системы менеджмента качества. Требования
(ISO 9001:2000) (Quality management systems. Requirements)
[4] ИСО/ТО 14969:1999 Системы качества. Изделия медицинские. Руководство по применению ИСО 13485
(ISO/TR 14969:1999) (Medical devices. Quality management systems. Guidance on the application of ISO 13485)
[5] ИСО 14971:2000 Медицинские изделия. Применение анализа риска к медицинским изделиям
(ISO 14971:2000) (Medical devices. Application of risk management to medical devices)
[6] ИСО 10012:2003 Системы управления измерениями. Требования к процессам измерения и измерительному оборудованию
(ISO 10012:2003) (Measurement management systems. Requirements for measurement processes and measuring equipment)
[7] ИСО 19011:2002 Руководящие указания по вопросам аудита систем менеджмента качества и/или систем менеджмента окружающей человека производственной среды
(ISO 19011:2002) (Guidelines for quality and/or environmental management systems auditing)
[8] ИСО 9004:2000 Системы менеджмента качества. Рекомендации по улучшению деятельности
(ISO 9004:2000) (Quality management systems. Guidelines for performance improvements)
[9] ИСО 14001:1996 Системы управления окружающей средой. Требования и руководство по применению
(ISO 14001:1996) (Environmental management systems. Specification with guidance for use)
Ключевые слова: система менеджмента качества, медицинские изделия, установленные требования, организация, документированная процедура, потребитель, верификация, валидация, политика и цели в области качества