4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
ОПРЕДЕЛЕНИЕ ОСТАТОЧНЫХ КОЛИЧЕСТВ ПЕСТИЦИДОВ
В ПИЩЕВЫХ ПРОДУКТАХ, СЕЛЬСКОХОЗЯЙСТВЕННОМ
СЫРЬЕ И ОБЪЕКТАХ ОКРУЖАЮЩЕЙ СРЕДЫ
Определение остаточных
количеств
квизалофоп-П-тефурила
по его основному метаболиту
квизалофоп-свободной кислоте в воде, почве, в семенах
и масле льна, сои, подсолнечника и в соломке льна
методом газожидкостной хроматографии
СБОРНИК МЕТОДИЧЕСКИХ УКАЗАНИЙ
МУК 4.1.1137-02
ВЫПУСК 1
МОСКВА
2004
1. Сборник подготовлен: Федеральным научным центром гигиены им. Ф.Ф. Эрисмана (чл.-корр. РАМН, проф. В.Н. Ракитский, проф. Т.В. Юдина); Московской сельскохозяйственной академией им. К.А. Тимирязева (проф. В.А. Калинин, к. хим. н. Довгилевич А.В.); Всероссийским НИИ фитопатологии (А.М. Макеев и др.); Всероссийским НИИ защиты растений (В.И. Долженко и др.); Санкт-Петербургским НИИ лесного хозяйства (Маслаков С.Е., Л.В. Григорьева и др.), при участии Департамента Госсанэпиднадзора Минздрава России (А.П. Веселов).
2. Методические указания рекомендованы к утверждению Комиссией по государственному санитарно-эпидемиологическому нормированию при Минздраве России.
3. Утверждены и введены в действие Главным государственным санитарным врачом Российской Федерации, Первым заместителем Министра здравоохранения Российской Федерации, академиком РАМН Г.Г. Онищенко.
4. Введены впервые.
Главный государственный санитарный
врач Российской Федерации, Первый
заместитель Министра здравоохранения
Российской Федерации
Г.Г. Онищенко
Дата введения: 1 января 2003 г.
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение остаточных количеств
квизалофоп-П-тефурила по его
основному метаболиту
квизалофоп-свободной кислоте в воде, почве, в семенах
и масле льна, сои, подсолнечника и в соломке льна
методом газожидкостной хроматографии
Методические указания
МУК 4.1.1137-02
1. Вводная часть
Фирма производитель: Юнироял Кемикал Ко. Инк.
Торговое наименование препарата: ПАНТЕРА, КЭ.
Действующее вещество (д. в.): квизалофоп-П-тефурил.
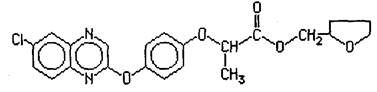
Название д. в. по номенклатуре ИЮПАК: (R)-2-[4-(6-=хлорхиноксалин-2-илокси)фенокси]пропионовой кислоты тетрагидрофурфуриловый эфир.
C22H21N2ClO5 M. м. 428
Кристаллический порошок белого цвета без запаха.
Давление паров: < 5,9 × 10-8 мм Hg при 25 °С.
Растворимость (мг/л при 25 °С): в воде - 3,753, метаноле - 6439, толуоле - 65170, н-гексан - 1243.
Температура плавления: 72,5 - 74,5 °С.
Коэффициент распределения в системе октанол/вода: logp - 4,3222.
Основным продуктом гидролитического разложения (метаболитом) квизалофоп-П-тефурила является 2-[4-(6-хлорхиноксалин-2-илокси) фенокси]пропионовая кислота.
Токсикологическая характеристика: ДСД для массы тела человека 0,004 мг/кг.
Гигиенические нормативы
ОДК для почвы - 0,1 мг/кг;
ОДУ для воды водоемов - 0,002 мг/дм3;
МДУ в семенах подсолнечника и сои - 0,04 мг/кг;
МДУ в масле подсолнечника и сои - 0,06 мг/кг.
ПАНТЕРА, КЭ - послевсходовый гербицид для борьбы с однолетними и многолетними злаковыми сорными растениями при возделывании овощных и технических культур.
2. Методика определения остаточных количеств квизалофоп-П-тефурила по его основному метаболиту квизалофоп-свободной кислоте в воде, почве, в семенах и масле льна, сои, подсолнечника и в соломке льна с применением газожидкостной хроматографии
2.1. Основные положения
2.1.1. Принцип метода
Метод основан на количественном определении квизалофоп-свободной кислоты - основного метаболита (продукта гидролиза) квизалофоп-П-тефурила. Метод включает извлечение квизалофоп-свободной кислоты и остаточных количеств квизалофоп-П-тефурила из анализируемого объекта органическим растворителем, проведение щелочного гидролиза квизалофоп-П-тефурила до квизалофоп-свободной кислоты, очистку пробы перераспределением веществ в системе несмешивающихся растворителей, дериватизацию квизалофоп-свободной кислоты (метилирование диазометаном) до квизалофоп-П-метила и очистку экстракта на колонке с оксидом алюминия.
Количественное определение проводят методом внешнего стандарта с применением газожидкостной хроматографии (ГЖХ) и использованием детектора постоянной скорости рекомбинации ионов (ДПР), детектора по захвату электронов (ДЭЗ) или термоионного детектора (ТИД), насадочной или капиллярной колонки с неподвижными фазами типа ОV-17 или SЕ-30.
2.1.2. Избирательность метода
Прочие пестициды, применяемые в интенсивной сельскохозяйственной технологии, определению не мешают. Очистка экстрактов, а также использование селективных детекторов позволяют устранить влияние мешающих анализу примесей.
2.1.3. Метрологическая характеристика метода
Метрологическая характеристика метода представлена в табл. 1, 2, 3.
Метрологическая характеристика метода
|
Анализируемый объект |
|||
|
масло |
семена |
соломка льна |
|
|
Предел обнаружения, мг/кг |
0,05 |
0,02 |
0,02 |
|
Диапазон определяемых концентраций, мг/кг |
0,05 - 0,5 |
0,02 - 0,5 |
0,02 - 0,5 |
|
Среднее значение определения, % |
79,3 |
86,7 |
81,0 |
|
Стандартное отклонение S, % |
11,7 |
11,4 |
12,2 |
|
Относительное стандартное отклонение DS, % |
6,5 |
6,3 |
6,8 |
|
Доверительный интервал среднего результата, % |
79,3 ± 13,9 |
86,7 ± 13,4 |
81,0 ± 14,6 |
Доверительный интервал и полнота определения
|
р = 0,95, п = 5 |
|||
|
Добавлено, мг/кг |
Обнаружено, % |
Доверительный интервал, ± % |
|
|
Масло |
0,05 |
72 |
15,9 |
|
0,1 |
81 |
14,8 |
|
|
0,5 |
85 |
13,0 |
|
|
Семена |
0,02 |
80 |
15,7 |
|
0,05 |
85 |
14,4 |
|
|
0,1 |
95 |
12,1 |
|
|
Соломка льна |
0,02 |
75 |
15,7 |
|
0,05 |
82 |
15,1 |
|
|
0,1 |
86 |
14,5 |
|
Полнота определения квизалофоп-П-тефурила в молельных пробах воды и почвы (р = 0,95, п = 6)
|
Внесено, мг/дм3, мг/кг |
Извлечено, % |
Доверительный интервал среднего результата, % |
|
|
Вода |
0,001 |
77,3 |
± 4,6 |
|
0,002 |
82,6 |
± 4,1 |
|
|
0,004 |
85,8 |
± 3,5 |
|
|
0,008 |
88,7 |
± 3,2 |
|
|
Почва |
0,05 |
74,5 |
± 5,3 |
|
0,1 |
78,2 |
± 4,9 |
|
|
0,2 |
82,7 |
± 4,4 |
|
|
0,4 |
85,5 |
± 3,8 |
2.2. Реактивы, растворы, материалы и оборудование
2.2.1. Реактивы, материалы и растворы
|
Аналитический стандарт квизалофоп-свободной кислоты |
|
|
Аналитический стандарт квизалофоп-П-метила |
|
|
Азот газообразный, высокой чистоты |
ТУ 301-07-25-89 |
|
Ацетон, осч |
ТУ 2633-004-11291058-94 |
|
Ацетонитрил, хч |
ТУ 6-09-4326-76 |
|
Вода дистиллированная и перегнанная над KMnO4 и щелочью |
|
|
Водород газообразный, высокой чистоты |
ТУ 301-07-27-90 |
|
н-Гексан, хч |
ТУ 6-09-3375-78 |
|
Гелий газообразный (сжатый) очищенный марки «А» |
ТУ 51-940-80 |
|
Диэтиловый эфир, ч |
|
|
Кислота серная, осч |
|
|
Кислота соляная, осч 23-4 |
ТУ 6-09-01-794-90 |
|
Калия гидроксид, чда |
|
|
Натрий серно-кислый безводный, чда |
|
|
Натрий хлористый, чда |
|
|
N-нитрозометилмочевина |
ТУ 6-09-11-1643-82 |
|
Оксид алюминия для хроматографии, ч, размер частиц 100 - 200 мкм, 1-й степени активности по Брокману, нейтральный |
|
|
Стандартные растворы квилазофоп-П-тефурила в ацетоне с концентрацией 0,05 и 0,5 мг/мл для фортификации проб |
|
|
Стандартные растворы квизалофоп-свободной кислоты в ацетоне с концентрациями 0,2; 0,5; 1,0; 2,0; 4,0; 8,0 мкг/мл для построения калибровочных графиков |
|
|
Стандартные растворы квизалофоп-П-метила в н-гексане с концентрацией 0,05; 0,5 мг/мл |
2.2.2. Приборы, аппаратура, посуда
|
Колонка хроматографическая набивная стеклянная длиной 1,0 м и внутренним диаметром 4,0 мм с неподвижной фазой 5 % OV-17 на Inerton Super-AW, 0,16 - 0,20 мм |
|
|
Колонка хроматографическая капиллярная кварцевая длиной 15 м и внутренним диаметром 0,53 мм с неподвижной фазой NBW-30 (типа OV-101) толщиной 1 мкм |
|
|
Колонка хроматографическая набивная стеклянная длиной 1,0 м и внутренним диаметром 3,0 мм с неподвижной фазой 5 % SE-30 на хроматоне N-Super, 0,125 - 0,160 мм |
|
|
Колонка хроматографическая капиллярная кварцевая длиной 5 м и внутренним диаметром 0,53 мм с неподвижной фазой НР-1 (типа SE-30) толщиной 2,65 мкм |
|
|
Колонки хроматографические стеклянные длиной 300 мм и внутренним диаметром 10 мм для очистки экстрактов |
|
|
Аппарат для встряхивания или аналогичный |
ТУ 64-1-1081-73 |
|
Баня водяная |
ТУ 64-1-2850-76 |
|
Весы аналитические типа ВЛА-200 |
ГОСТ 34104-80 |
|
Весы лабораторные типа ВЛКТ-500 |
ГОСТ 24104-80 |
|
Пробирки мерные конические на 10 мл |
ГОСТ 1770-7420 |
|
Воронка Бюхнера |
|
|
Воронки делительные емкостью 500 и 1000 мл |
|
|
Воронки для фильтрования стеклянные |
|
|
Колба Бунзена емкостью 250 мл |
|
|
Колбы круглодонные, емкостью 250 мл |
|
|
Колбы мерные, емкостью 250, 1000 мл |
|
|
Колбы плоскодонные на 250, 500 мл |
|
|
Колбы мерные, емкостью 10, 50, 100 мл |
|
|
Концентраторы грушевидные (конические) НШ 19, КГУ-100-14/19, ТС |
ГОСТ 10394 |
|
Мельница лабораторная (кофемолка) |
|
|
Микрошприц МШ 10А |
ТУ 64-1-2850 |
|
Насос стеклянный вакуумный водоструйный |
ГОСТ 10696-75 |
|
Пипетки мерные, емкостью 0,1, 1, 2, 5 и 10 мл |
|
|
Ротационный испаритель ИР-1М или аналогичного типа |
ТУ 25-11-917-76 |
|
Сито с диаметром отверстий 1,0 мм |
|
|
Стаканы стеклянные на 100 мл |
ГОСТ 6236 |
|
Термометр градуировочный (при погружении всей нижней части) |
ГОСТ 2823-73 |
|
Универсальная индикаторная бумага |
ТУ 6-09-1181-76 |
|
Установка для перегонки растворителей при атмосферном давлении |
|
|
Установка для упаривания органических растворителей в токе азота |
|
|
Установка ультразвуковая (УЗ-баня) «Серьга» УЗМ 002 или другая аналогичного типа. Выходная электрическая мощность генератора не менее 70 Вт; рабочая частота установки - 22 кГц ± 7,5 % |
|
|
Фильтры бумажные, «красная лента» |
ТУ 6-09-1678-86 |
|
Фильтры бумажные, «белая лента» |
ТУ 6-09-1678-86 |
|
Фильтры бумажные, «синяя лента» |
ТУ 6-09-1678-86 |
|
Фильтр Шотта IV |
ГОСТ 9775-61 |
|
Флаконы стеклянные (типа пенициллиновых), емкостью 5 и 10 мл |
|
|
Чашка фарфоровая |
2.3. Подготовка к определению
2.3.1. Подготовка и очистка реактивов и растворителей
Перед началом работы рекомендуется проверить чистоту применяемых органических растворителей. Для этого 100 мл растворителя упаривают в ротационном испарителе при температуре 40 °С до объема 1,0 мл и хроматографируют. При обнаружении мешающих определению примесей проводят очистку растворителей в соответствии с общепринятыми методиками.
2.3.2. Подготовка и кондиционирование хроматографической набивной колонки
Подготовленной насадкой заполняют стеклянную колонку и уплотняют ее под вакуумом. Колонку устанавливают в термостате хроматографа, не подсоединяя к детектору, и кондиционируют ее при рабочем расходе газа-носителя и температуре 280 °С в течение 8 - 10 ч.
2.3.3. Приготовление стандартных растворов
2.3.3.1. Стандартные растворы квизалофоп-П-метила. Для приготовления маточного стандартного раствора навеску квизалофоп-П-метила массой 0,1 г растворяют в гексане в мерной колбе емкостью 200 мл. После растворения вещества раствор в колбе доводят до метки гексаном. Полученный раствор соответствует концентрации 0,5 мг/мл. Далее 1,0 мл гексанового раствора с концентрацией 0,5 мг/мл помещают в мерную колбу емкостью 100 мл и доводят гексаном до метки. Полученный раствор имеет концентрацию 5,0 мкг/мл.
Мерные колбы закрывают пробками и растворы тщательно перемешивают. Срок годности растворов не более 0,5 года при условии хранения в холодильнике.
Данные растворы используют для определения времени удерживания квизалофоп-П-метила в целях дальнейшей идентификации вещества в анализируемых пробах и отработки условий хроматографирования.
При изучении полноты определения квизалофоп-П-тефурила и квизалофоп-свободной кислоты для фортификации модельных проб используют ацетоновые растворы квизалофоп-П-тефурила с концентрацией 0,05 или 0,5 мг/мл.
2.3.3.2. Стандартные растворы квизалофоп-свободной кислоты. Для приготовления маточного стандартного раствора квизалофоп-свободной кислоты навеску массой 0,1 г растворяют в ацетоне в мерной колбе емкостью 200 мл. После растворения вещества раствор в колбе доводят до метки ацетоном. Полученный раствор соответствует концентрации 0,5 мг/мл. Далее 1,0 мл ацетонового раствора с концентрацией 0,5 мг/мл помещают в мерную колбу емкостью 100 мл и доводят ацетоном до метки. Полученный раствор имеет концентрацию 5,0 мкг/мл. Из этого раствора получают методом дальнейших последовательных разведений ацетоном растворы следующих концентраций: 0,2; 0,5; 1,0; 2,0; 4,0; 8,0 мкг/мл.
Мерные колбы закрывают пробками и растворы тщательно перемешивают. Срок годности растворов не более 0,5 года при условии хранения в холодильнике.
Полученные растворы используют для построения калибровочных графиков.
2.3.4. Построение калибровочных графиков
Для построения калибровочного графика осуществляется следующая процедура: по 1,0 мл приготовленных ацетоновых растворов квизалофоп-свободной кислоты (см. п. 2.3.3.2) упаривают досуха в токе азота при комнатной температуре и далее переходят к стадиям метилирования (п. 2.5.4) и очистки (п. 2.5.5). В результате получают растворы квизалофоп-П-метила различных концентраций в 1,0 мл гексана.
Осуществляют не менее 5 параллельных измерений и находят среднее значение высоты (площади) хроматографического пика для каждой концентрации. Строят калибровочный график зависимости высоты (площади) хроматографического пика от концентрации квизалофоп-свободной кислоты.
2.3.5. Подготовка и кондиционирование хроматографических колонок с оксидом алюминия 1 степени активности по Брокману для очистки экстрактов
Оксид алюминия прогревают в фарфоровой чашке в термостате при 150 °С в течение 5 ч и охлаждают до комнатной температуры (1 степень активности по Брокману). Колбу с оксидом алюминия хранят герметично закрытой. Нижний зауженный конец стеклянной колонки закрывают тампоном из хлопковой ваты и заполняют (при легком встряхивании) 5,0 г оксида алюминия (высота слоя ~ 7,0 см). Над слоем оксида алюминия помещают слой безводного сульфата натрия высотой 2,0 см и перед использованием промывают колонку 20,0 мл гексана.
2.3.6. Приготовление эфирного раствора диазометана
В пенициллиновый флакон емкостью 5,0 мл помещают навеску нитрозометил-мочевины 100 - 120 мг и флакон пломбируют резиновой пробкой. В другой флакон такого же объема наливают 2,0 мл диэтилового эфира. Флакон пломбируют резиновой пробкой и охлаждают в морозильной камере в течение 1 ч при температуре 8 - 10 °С. Затем готовят установку для получения диазометана (схема установки представлена на рис. 6). Резиновую пробку флакона с эфиром прокалывают медицинской иглой диаметром 1,0 мм таким образом, чтобы один конец находился над поверхностью эфира, а другой выходил наружу (для сброса излишнего давления диазометана). Два флакона после этого соединяют между собой гибкой полихлорвиниловой трубкой-перемычкой, на концах которой герметично вставлены медицинские иглы диаметром 2,0 мм. Одна игла находится в свободном объеме флакона с нитрозометилмочевиной, другая игла погружается в слой эфира на дно флакона-приемника. Затем пробка флакона с нитрозометилмочевиной прокалывается иглой медицинского шприца на 1,0 мл, и из шприца по каплям вносится 50 %-ный водный раствор гидроокиси калия. Образовавшийся в ходе реакции диазометан пропускается через эфир до насыщения (появление желтизны спелого лимона). Процедуру обязательно проводят в работающем вытяжном шкафу.
2.4. Отбор проб
Отбор проб для анализа проводят в соответствии с «Унифицированными правилами отбора проб сельскохозяйственной продукции, продуктов питания и объектов окружающей среды для определения микроколичеств пестицидов», утвержденными 21.08.79 № 2051-79.
Пробы воды при наличии взвеси фильтруют через бумажный фильтр белая лента и хранят в закрытой стеклянной таре при температуре 4 - 6 °С не более 3 дней.
Пробы почвы просушивают до воздушно-сухого состояния при комнатной температуре в отсутствии прямого солнечного света и хранят в закрытой стеклянной или полиэтиленовой таре.
Семена и соломку просушивают до воздушно-сухого состояния и хранят до анализа в закрытой стеклянной или полиэтиленовой таре.
Пробы почвы перед анализом рассыпают на бумаге или кальке и пестиком разминают крупные комки, из проб пинцетом удаляют включения: корни растений, насекомых, камни, стекло, кости, уголь и другие.
После этого пробы почвы растирают в ступке пестиком, просеивают через сито с диаметром отверстий 1,0 мм и после перемешивания отбирают усредненную аналитическую пробу.
Перед анализом сухие пробы измельчают: семена - в лабораторной мельнице (кофемолке), соломку - ножницами и после перемешивания отбирают усредненную аналитическую пробу.
2.5. Описание определения
2.5.1. Вода
Аналитическую пробу воды объемом 500 см3 помещают в делительную воронку емкостью 1000 мл и добавляют 5,0 мл 5 %-ного водного раствора гидроокиси калия. Содержимое воронки перемешивают встряхиванием и оставляют на 60 мин при температуре 4 - 6 °С. После этого в воронку добавляют 100 мл насыщенного водного раствора хлористого натрия. Содержимое воронки перемешивают, добавляют 100 мл н-гексана и энергично встряхивают в течение 2 мин. После 5-минутного отстаивания нижний водно-солевой слой сливают в мерную колбу емкостью 1000 мл, а верхний гексановый слой отбрасывают. Водно-солевую фракцию возвращают в делительную воронку и процедуру очистки пробы повторяют еще раз с использованием 50 мл гексана. Делительную воронку промывают водопроводной и ополаскивают дистиллированной водой.
В водно-солевую фракцию, находящуюся в колбе, добавляют при постоянном перемешивании по каплям концентрированную серную кислоту до уровня рН 1 - 2. Водно-солевую фракцию помещают в делительную воронку и добавляют 75 мл смеси гексан-диэтиловый эфир (80 : 20, по объему). Содержимое воронки энергично встряхивают в течение 2 мин. После 5-минутного отстаивания нижний водно-солевой слой сливают в мерную колбу емкостью 1000 мл, а верхний гексано-эфирный слой фильтруют через слой безводного сульфата натрия (толщина слоя - 1,0 - 1,5 см), помещенный в воронке для фильтрования на бумажный фильтр «синяя лента», в круглодонную колбу емкостью 250 мл. Экстрагирование и фильтрование повторяют еще два раза с использованием последовательно 75 и 50 мл смеси гексан-диэтиловый эфир (80 : 20, по объему). После этого водно-солевую фракцию отбрасывают.
Объединенный гексано-эфирный экстракт, находящийся в колбе, помещают в ротационный вакуумный испаритель и упаривают растворители досуха при температуре 40 °С. Во флакон после его охлаждения до комнатной температуры вносят 1 мл свежеприготовленного эфирного раствора диазометана (п. 2.3.6), флакон закрывают пробкой и оставляют при комнатной температуре на 2 ч. После этого в работающем вытяжном шкафу в токе азота при комнатной температуре во флаконе упаривают досуха диэтиловый эфир и остаточные концентрации диазометана. Во флакон с сухим остатком вносят 0,5 мл ацетона и проводят количественное определение квизалофоп-П-тефурила по п. 2.6.
2.5.2. Почва
Аналитическую пробу почвы массой 20,0 ± 0,1 г помещают в плоскодонную колбу емкостью 250 мл, добавляют 50 мл 1,0 %-ного водного раствора гидроокиси калия и 50 мл ацетона. Содержимое колбы перемешивают встряхиванием и подвергают обработке ультразвуком в УЗ-бане по 5 мин два раза. Колбу с пробой закрывают и оставляют на 14 - 16 ч (на ночь) при комнатной температуре. После этого содержимое колбы встряхивают, подвергают обработке ультразвуком в УЗ-бане по 5 мин два раза и фильтруют через бумажный фильтр «белая лента» под вакуумом на воронке Бюхнера в колбу Бунзена емкостью 250 мл. Оставшуюся часть пробы в колбе промывают 20 мл ацетона и экстракт также фильтруют.
При использовании аппарата для встряхивания в плоскодонную колбу с аналитической пробой вносят 50 мл 1,0 %-ного водного раствора гидроокиси калия и 50 мл ацетона и встряхивают в течение 60 мин. Колбу с пробой закрывают и оставляют на 14 - 16 ч (на ночь) при комнатной температуре. После этого содержимое колбы встряхивают 30 мин с использованием аппарата для встряхивания и фильтруют через бумажный фильтр «белая лента» под вакуумом на воронке Бюхнера в колбу Бунзена емкостью 250 мл. Оставшуюся часть пробы в колбе промывают 20 мл ацетона и экстракт также фильтруют.
Объединенный экстракт количественно переносят в делительную воронку емкостью 500 мл, добавляют 50 мл насыщенного водного раствора хлористого натрия. Содержимое воронки перемешивают, добавляют 50 мл гексана и энергично встряхивают в течение 2 мин. После 5-минутного отстаивания нижний слой сливают в мерную колбу емкостью 250 мл, а верхний гексановый слой отбрасывают. Водно-солевую фракцию возвращают в делительную воронку и процедуру очистки экстракта повторяют еще раз с использованием 50 мл гексана. Делительную воронку промывают водопроводной и ополаскивают дистиллированной водой.
В водно-солевую фракцию, находящуюся в колбе, добавляют при постоянном перемешивании по каплям концентрированную серную кислоту до уровня рН 1 - 2. Водно-солевую фракцию переносят в делительную воронку и добавляют 50 мл смеси гексан-диэтиловый эфир (80 : 20, по объему). Содержимое воронки энергично встряхивают в течение 2 мин. После 5-минутного отстаивания нижний слой сливают в мерную колбу емкостью 250 мл, а верхний гексано-эфирный слой фильтруют через слой безводного сульфата натрия (толщина слоя - 1,0 - 1,5 см), помещенный в воронке для фильтрования на бумажный фильтр «синяя лента», в круглодонную колбу емкостью 250 мл. Экстрагирование и фильтрование повторяют еще два раза с использованием последовательно 50 и 30 мл смеси гексан-диэтиловый эфир (80 : 20, по объему). После этого водно-солевую фракцию отбрасывают.
Колбу с объединенным гексано-эфирным экстрактом помещают в ротационный вакуумный испаритель и упаривают растворители досуха при температуре 40 °С. Во флакон после его охлаждения до комнатной температуры вносят 1 мл свежеприготовленного эфирного раствора диазометана (п. 2.3.6), флакон закрывают пробкой и оставляют при комнатной температуре на 2 ч. После этого в работающем вытяжном шкафу в токе азота при комнатной температуре во флаконе упаривают досуха диэтиловый эфир и остаточные концентрации диазометана. Сухой остаток растворяют в 1 мл ацетона и проводят количественное определение квизалофоп-П-тефурила по п. 2.6.
2.5.3. Семена, масло и соломка льна
2.5.3.1. Экстракция семян. Навеску измельченных семян массой 10,0 ± 0,1 г помещают в плоскодонную колбу емкостью 250 мл, заливают 100 мл ацетона и подвергают обработке ультразвуком в УЗ-бане в течение 5 мин. Затем для отделения механических примесей от образовавшейся суспензии экстракт пропускают через коническую воронку с хлопковой ватой в химический стакан. Воронку и вату промывают 10 мл ацетона. Далее экстракт фильтруют с помощью воронки Бюхнера (фильтр «белая лента») с последующей промывкой фильтра 10 мл ацетона. Экстракт переносят в делительную воронку емкостью 500 мл, добавляют 100 мл 0,5 %-ного водного раствора гидроокиси калия и выдерживают смесь в течение 1 ч.
Далее в воронку добавляют 150 мл насыщенного раствора хлорида натрия, перемешивают и добавляют 50 мл гексана. Воронку встряхивают в течение 2 мин и после разделения слоев нижний водный слой собирают в колбу, а гексановый слой отбрасывают. Делительную воронку тщательно промывают дистиллированной водой и переносят в нее водную фракцию, добавляют 50 мл гексана и процедуру повторяют еще раз. Водный слой собирают в плоскодонную колбу емкостью 500 мл и подкисляют 2,0 - 2,5 мл концентрированной HCl (до рН 1,0 - 2,0), контролируя рН с помощью индикаторной бумаги, и пропускают через фильтр Шотта № 4. Затем водную фракцию переносят в чистую делительную воронку емкостью 500 мл и добавляют 100 мл смеси гексан-диэтиловый эфир (1 : 1). Воронку встряхивают в течение минуты и после разделения слоев (выдерживать не менее 30 мин) водный слой отбрасывают. В случае образования эмульсии в оставшийся органический слой добавляют несколько капель этилового спирта, осторожно перемешивают и пропускают через коническую воронку с ватой и слоем безводного сульфата натрия (толщина 1,0 - 1,5 см) в грушевидную колбу емкостью 200 мл. Воронку и сульфат натрия промывают 5 мл гексана. Объединенный экстракт упаривают на ротационном вакуумном испарителе при температуре водяной бани 40 °С до объема 5 - 10 мл. Остаток экстракта переносят в коническую колбу или пенициллиновый флакон и упаривают досуха в токе азота при температуре 60 °С. Дальнейшие метилирование и очистку экстракта проводят в соответствии с п. п. 2.5.4 и 2.5.5.
2.5.3.2. Экстракция масла. Образец масла массой 5,0 ± 0,1 г помещают в делительную воронку емкостью 500 мл, добавляют 120 мл гексана и хорошо перемешивают. Затем в делительную воронку добавляют 200 мл ацетонитрила, воронку интенсивно встряхивают в течение 1 мин и выдерживают смесь не менее 30 мин. После разделения слоев нижний ацетонитрильный слой переносят в делительную воронку емкостью 1000 мл, добавляют 180 мл 0,5 %-ного раствора гидроокиси калия и выдерживают смесь 1 ч при комнатной температуре. Затем в делительную воронку добавляют 100 мл насыщенного раствора хлорида натрия и хорошо перемешивают. Далее в делительную воронку добавляют 100 мл гексана, интенсивно встряхивают в течение 2 мин и после разделения слоев нижний водный слой сливают в плоскодонную колбу емкостью 500 мл, гексановый слой отбрасывают. Делительную воронку тщательно промывают, водную фракцию переносят в воронку и повторно проводят процедуру очистки с использованием 100 мл гексана.
Далее водную фракцию подкисляют 3,0 - 3,5 мл концентрированной HCl (до рН 1,0 - 2,0). Делительную воронку тщательно промывают и переносят в нее подкисленную водную фракцию. В делительную воронку добавляют 100 мл смеси гексан-диэтиловый эфир (1 : 1). Воронку интенсивно встряхивают в течение 2 мин и после разделения слоев нижний водный слой отбрасывают, а органическую фазу пропускают через коническую воронку с ватой и слоем безводного сульфата натрия (толщина 1,0 - 1,5 см) в грушевидную колбу емкостью 200 мл. Воронку и сульфат натрия промывают 5 мл гексана. Объединенный экстракт упаривают на ротационном испарителе при температуре водяной бани 40 °С до объема 5 - 10 мл. Остаток экстракта переносят в коническую колбу или пенициллиновый флакон и упаривают досуха в токе азота при температуре 60 °С. Дальнейшие метилирование и очистку экстракта проводят в соответствии с п. п. 2.5.4 и 2.5.5.
2.5.3.3. Экстракция соломки льна. Измельченный образец соломки льна массой 10,0 ± 0,1 г помещают в плоскодонную колбу емкостью 250 мл, заливают 150 мл ацетона и подвергают обработке ультразвуком в УЗ-бане в течение 5 мин. Затем экстракт отфильтровывают с помощью воронки Бюхнера (на фильтре «красная лента»). Фильтр промывают 10 мл ацетона. Экстракт переносят в делительную воронку емкостью 500 мл, добавляют 150 мл 0,5 %-ного водного раствора гидроокиси калия и выдерживают смесь в течение 1 ч при комнатной температуре. Далее добавляют 75 мл насыщенного раствора хлорида натрия, перемешивают и добавляют 50 мл гексана. Воронку встряхивают в течение 2 мин и после разделения слоев нижний водный слой собирают, а гексановый слой отбрасывают. Делительную воронку тщательно промывают дистиллированной водой и переносят в нее водную фракцию, добавляют 50 мл гексана и процедуру повторяют еще раз. Водный слой собирают в плоскодонную колбу емкостью 500 мл и подкисляют 2,5 - 3,0 мл концентрированной HCl (до рН 1,0 - 2,0). После этого водную фракцию переносят в чистую делительную воронку емкостью 500 мл и добавляют 100 мл смеси гексан-диэтиловый эфир (1 : 1). Воронку встряхивают в течение 2 мин и после разделения слоев водный слой отбрасывают, а органическую фазу пропускают через коническую воронку с ватой и слоем безводного сульфата натрия (толщина 1,0 - 1,5 см) в грушевидную колбу емкостью 200 мл. Воронку и сульфат натрия промывают 5 мл гексана. Объединенный экстракт упаривают на ротационном испарителе при температуре водяной бани 40 °С до объема 5 - 10 мл. Остаток экстракта переносят в коническую колбу или пенициллиновый флакон и упаривают досуха в токе азота при 60 °С. Дальнейшие метилирование и очистку экстракта проводят в соответствии с п. п. 2.5.4 и 2.5.5.
2.5.4. Метилирование
К сухому остатку (из п. п. 2.5.3.1, 2.5.3.2, 2.5.3.3) добавляют 1,0 мл эфира, насыщенного диазометаном, флакон закрывают и оставляют на 40 мин при комнатной температуре. Затем эфир упаривают в токе азота досуха, растворяют сухой остаток в 2,0 мл гексана и экстракт подвергают очистке по п. 2.5.5.
2.5.5. Очистка
Гексановый экстракт (из п. 2.5.4) количественно переносят на подготовленную колонку с оксидом алюминия 1-й степени активности по Брокману. После этого колонку промывают 20 мл н-гексана, который отбрасывают. Квизалофоп-П-метил элюируют 30 мл диэтилового эфира со скоростью не более 60 капель в минуту в грушевидную колбу-концентратор. Элюат упаривают в токе азота досуха при комнатной температуре. Сухой остаток растворяют в 1,0 мл гексана и анализируют на содержание квизалофоп-П-метила в соответствии с п. 2.6.
2.6. Условия газохроматографического анализа
2.6.1. Для набивной колонки с неподвижной фазой 5 % OV-17
|
Газовый хроматограф «Hewlett-Packard 5890» с ДЭЗ (порог чувствительности по линдану не более 3 × 10-10 мг/с) или газовый хроматограф «Цвет-500» с ДПР (порог чувствительности по линдану не более 5 × 10-10 мг/с) |
|
|
Колонка стеклянная спиральная длиной 1,0 м и внутренним диаметром 4,0 мм |
|
|
Носитель - Inerton Super-AW (0,16 - 0,20 мм) с неподвижной фазой 5 % OV-17 |
|
|
Рабочая шкала электрометра |
2 × 10-10А |
|
Скорость протяжки ленты самописца |
0,3 см/мин |
|
Скорость потока газа-носителя (азота в/ч) |
40 см3/мин |
|
Температура колонки |
280 °С |
|
испарителя |
290 °С |
|
детектора |
300 °С |
|
Объем вводимой пробы |
2,0 мкл |
|
Время удерживания квизалофоп-П-метила |
5,0 ± 0,1 мин |
|
Линейный диапазон определения |
0,2 - 20 нг |
После выполнения 5 - 7 анализов набивную колонку следует прогреть при температуре 300 °С в течение 1 ч.
2.6.2. Для набивной колонки с неподвижной фазой 5 % SE-30
|
Колонка хроматографическая набивная длиной 1 м и внутренним диаметром 3 мм с неподвижной фазой 5 % SE-30 на Хроматоне N-Super, 0,125 - 0,16 мм |
|
|
Показания электрометра |
32 × 1010 |
|
Скорость движения ленты самописца |
0,25 см/мин |
|
Температура испарителя |
260 °С |
|
колонки |
260 °С |
|
детектора |
310 °С |
|
Расход газа-носителя (азота в/ч) |
40 см3/мин |
|
Расход водорода и воздуха для ТИД |
15 и 300 см3/мин соответственно |
|
Объем вводимой пробы |
2 мкл |
|
Время удерживания квизалофоп-П-метила |
2,7 ± 0,2 мин |
|
Предел детектирования |
0,5 нг |
|
Линейный диапазон детектирования |
0,5 - 8,0 нг |
2.6.3. Для капиллярной колонки с неподвижной фазой NBW-30 (типа OV-101)
|
Газовый хроматограф «Hewlett-Packard 5890» с ДЭЗ (порог чувствительности по линдану не более 3 × 10-10 мг/с) или газовый хроматограф «Цвет-500» с ДПР (порог чувствительности по линдану не более 5 × 10-10 мг/с) |
|
|
Колонка кварцевая капиллярная длиной 5 м, внутренним диаметром 0,53 мм с неподвижной фазой NBW-30 (типа OV-101) толщиной 1 мкм |
|
|
Рабочая шкала электрометра |
2 × 10-10A |
|
Скорость потока азота в/ч |
10 см3/мин |
|
Скорость потока дополнительного газа (азота в/ч) к детектору |
40 см3/мин |
|
Температура колонки |
250 °С |
|
испарителя |
270 °С |
|
детектора |
300 °С |
|
Объем вводимой пробы |
2,0 мкл |
|
Время удерживания квизалофоп-П-метила |
3,0 ± 0,1 мин |
|
Линейный диапазон определения |
0,2 - 20 нг |
2.6.4. Для капиллярной колонки с неподвижной фазой SE-30
|
Колонка кварцевая капиллярная длиной 5 м, внутренним диаметром 0,53 мм с неподвижной фазой НР-1 (типа SE-30), 2,65 мкм |
|
|
Температура колонки |
программирование от 180 (3 мин) до 280 °С (15 мин) со скоростью 10 °С/мин |
|
испарителя |
260 °С |
|
детектора |
300 °С |
|
Расход газов: |
|
|
газа-носителя (гелий марки «А») |
7 см3/мин |
|
водорода и воздуха к ТИД |
30 и 300 см3/мин соответственно |
|
дополнительного газа (гелий марки «А») к ТИД |
60 см3/мин |
|
дополнительного газа (азот в/ч) к ДЭЗ |
40 см3/мин |
|
Объем вводимой пробы |
1 мкл |
|
Время удерживания квизалофоп-П-метила |
11,7 ± 0,1 мин |
|
Предел детектирования |
0,5 нг |
|
Линейный диапазон детектирования |
0,5 - 8,0 нг |
Регистрацию хроматограмм при работе на газовом хроматографе «Hewlett-Packard 5890» проводили на электронном интеграторе «HP 3396» (параметры интегрирования: порог обнаружения пиков (Thrsh) = 4, ширина пика (PkWd) = 0,04). При использовании газового хроматограф «Цвет-500» может быть применен самописец КСП-4 (при обработке результатов вручную) или компьютерная система обработки данных типа «Мультихром».
2.7. Обработка результатов анализа
Содержание квизалофоп-П-тефурила рассчитывают методом внешнего стандарта по формуле:
![]() ,
где
,
где
Х - содержание квизалофоп-П-тефурила в пробе, мг/кг или мг/дм3;
Н1 - высота (площадь) пика анализируемого вещества, мм (мм2);
Н0 - высота (площадь) пика стандартного раствора квизалофоп-П-метила, мм (мм2);
А - концентрация стандартного раствора квизалофоп-П-метила, мкг/мл;
V - объем экстракта, подготовленного для хроматографирования, мл;
M - объем (см3) или масса (г) аналитической пробы;
1,2 - коэффициент пересчета, учитывающий соотношение молекулярных масс квизалофоп-П-метила и квизалофоп-П-тефурила.
При получении достоверных результатов измерений конечный результат записывается следующим образом:
![]()
![]() ,
где
,
где
X - содержание остаточных количеств квизалофоп-П-тефурила в анализируемой пробе;
х - среднее из трех параллельных результатов измерений;
Δ - показатель погрешности измерений, соответствующий диапазону, в который попадает величина х.
3. Требования техники безопасности
Необходимо соблюдать общепринятые правила техники безопасности при работе с органическими растворителями, токсичными веществами, электронагревательными приборами и сжатыми газами, а также требования, изложенные в документации на приборы.
4. Разработчики
Долженко В.И., Крылов А.И., Николаев М.Я., Шеболдасова Н.М., Жаковская З.А., Тарарин П.А., Григорьева Л.В. (ВНИИ защиты растений, 189620, Санкт-Петербург - Пушкин, шоссе Подбельского, 3).
СОДЕРЖАНИЕ
|
2.2. Реактивы, растворы, материалы и оборудование. 3 2.3. Подготовка к определению.. 5 2.6. Условия газохроматографического анализа. 10 2.7. Обработка результатов анализа. 12 |