РОССИЙСКОЕ АКЦИОНЕРНОЕ ОБЩЕСТВО
ЭНЕРГЕТИКИ И ЭЛЕКТРИФИКАЦИИ
«ЕЭС РОССИИ»
Департамент научно-технической политики и развития
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
ПО КОНТРОЛЮ СОСТОЯНИЯ
ОСНОВНОГО ОБОРУДОВАНИЯ
ТЕПЛОВЫХ ЭЛЕКТРИЧЕСКИХ СТАНЦИЙ
ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА
И ХИМИЧЕСКОГО СОСТАВА ОТЛОЖЕНИЙ

CO 34.37.306-2001
(РД 153-34.1-37.306-2001)
ОАО «ВТИ»
Москва 2003
Разработано Открытым акционерным обществом «Всероссийский дважды ордена Трудового Красного Знамени теплотехнический научно-исследовательский институт» (ОАО «ВТИ»);
ОАО «Фирма по наладке, совершенствованию технологии и эксплуатации электростанций и сетей ОРГРЭС».
Исполнители Б.С. ФЕДОСЕЕВ, Н.М. КАЛИНИНА, Ю.В. КОЗЛОВ, Б.Э. ШКОЛЬНИКОВА, В.В. ХОЛЩЕВ.
Утверждено Департаментом научно-технической политики и развития РАО «ЕЭС России» 11 декабря 2001 г.
Первый заместитель начальника А.Л. ЛИВИНСКИЙ
Срок первой проверки настоящего СО - 2007 г.,
периодичность проверки - один раз в 5 лет.
Ключевые слова: паровые турбины, экранные трубы, коллекторы, барабаны паровых котлов, отложения, химический анализ отложений, контрольные образцы труб.
|
МЕТОДИЧЕСКИЕ УКАЗАНИЯ ПО КОНТРОЛЮ СОСТОЯНИЯ ОСНОВНОГО ОБОРУДОВАНИЯ ТЕПЛОВЫХ ЭЛЕКТРОСТАНЦИЙ ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА И ХИМИЧЕСКОГО СОСТАВА ОТЛОЖЕНИЙ |
СО 34.37.306-2001 (РД 153-34.1-37.306-2001) Взамен РД 34.37.306-87 |
Срок действия установлен
с 2002-01-01
до 2012-01-01
Настоящий стандарт организации устанавливает порядок осмотра состояния поверхностей нагрева котлов и проточной части паровых турбин, способы отбора проб, методы определения количества и химического состава отложений, образовавшихся в проточной части турбин и на поверхностях нагрева котлов тепловых электростанций.
1 ОБЩИЕ ПОЛОЖЕНИЯ
Контроль за состоянием теплосилового оборудования осуществляется путем осмотра проточной части паровых турбин, экранных труб, коллекторов и барабанов паровых котлов.
Осмотры котлов и их элементов, а также паровых турбин для оценки коррозионного состояния металла и солевых загрязнений проводятся во время капитальных ремонтов.
Все операции по осмотру оборудования должны выполняться представителями химического и котлотурбинного цехов.
2 ОЦЕНКА СОСТОЯНИЯ ПРОТОЧНОЙ ЧАСТИ ТУРБИНЫ
Для оценки степени загрязнения проточной части турбины необходимо регулярно вести наблюдения за давлением пара в контрольных ступенях турбины (в камере регулирующей ступени, в межкорпусном пространстве двухцилиндровых турбин и отборах) при нагрузках 80 - 100 % номинальной. Давление измеряют манометрами класса точности 0,6 - 1,0.
По данным наблюдений строят график изменения давления по времени в зависимости от нагрузки, который показывает динамику роста давлений в контрольных ступенях в зависимости от износа проточной части турбины. Значения давлений в контрольных ступенях необходимо сопоставлять при одинаковых расходах пара. Повышение давления в контрольных ступенях по сравнению с номинальным при данном расходе пара должно быть не более 10 %. При этом оно не должно превышать предельных значений, установленных заводом-изготовителем.
При достижении предельных значений должна быть проведена промывка или очистка проточной части турбины. Способ промывки или очистки должен быть выбран исходя из состава и характера отложений и местных условий. Возможные варианты: промывка влажным паром, механическая очистка, химическая промывка.
2.1 Отбор проб
Пробы отложений из проточной части турбины должны быть отобраны по возможности сразу после вскрытия цилиндра турбины, но не позднее первых суток.
При отборе проб оценивают количество, расположение, цвет, плотность, прочность связи с металлом и прочие характерные особенности отложений, а также коррозионные и эрозионные повреждения металла. Результаты записывают в журнал осмотров теплосилового оборудования. Отбор проб отложений выполняют при помощи скальпеля, перочинного ножа или особых скребочков, изготовленных из обычной стали в форме ложки по профилю лопаточного аппарата каждой ступени или ряда ступеней. При снятии отложений под очищаемую лопатку подставляют кусочек кальки или глянцевой бумаги, согнутой желобком. Снятые отложения по возможности без потерь ссыпают в пакетики из кальки или в предварительно высушенные и взвешенные бюксы, причем в каждый бюкс помещают отложения только с одной ступени. Пакеты из кальки с пробами хранят в эксикаторе с прокаленной СаО. В журнале осмотра отмечают количество лопаток, отложения с которых помещены в данный бюкс.
Выбранные лопатки по возможности полностью освобождают от отложений. Это необходимо для подсчета общего количества отложений на каждой ступени и во всей проточной части турбины. Отложения, снятые с бандажей и направляющих лопаток, учитывают и исследуют отдельно.
Пробы следует отбирать с нескольких лопаток в количестве не менее 0,5 г. Отобранные пробы переносят в лабораторию и взвешивают.
Данные заносят в журнал осмотра теплосилового оборудования (таблица 1).
Таблица 1
Журнал осмотра теплосилового оборудования
|
Количество лопаток на данной ступени |
Количество очищенных лопаток |
Вес снятых отложений со всех очищенных лопаток, г |
Среднее количество отложений на одну лопатку, г |
Количество отложений на одной ступени, г |
Количество отложений по цилиндрам, г |
|||
|
ЦВД |
ЦСД |
ЦНД |
||||||
|
|
|
|
|
|
|
|
|
|
2.2 Подготовка к анализу
Если пробу разделяют для выполнения параллельных анализов в лабораториях электростанции и химической службы, то необходимо обеспечить ее однородность. Для этого каждую пробу, подвергающуюся делению, предварительно тщательно измельчают и перемешивают.
Результаты химического анализа отложений, выполненные лабораторией химической службы энергосистемы, с соответствующим заключением направляют на имя главного инженера электростанции и присоединяют ко всей документации по осмотру данной турбины.
2.3 Определение объема химического анализа отложений
Объем химического анализа отложений определяется конкретно поставленной задачей (характеристикой и оценкой водно-химического режима, выбором способа промывки проточной части и пр.). Если количество отложений не более 0,2 г, то определяют только содержание наиболее важных компонентов - кремнекислоты, железа, меди и натрия.
2.4 Методика динамического анализа отложений
Отложения, образующиеся в проточной части паровых турбин, могут содержать соединения натрия, кальция и магния, железа, меди, цинка, реже другие металлы. Присутствие соединений натрия и щелочноземельных металлов свидетельствует о загрязнении пара этими веществами. Оксиды металлов являются продуктами коррозии или эрозии конструкционных материалов. При анализе следует отдельно подвергать исследованию водорастворимую и нерастворимую в воде части отложений. В водорастворимой части определяют содержание натрия, кальция и магния, хлоридов, сульфатов, фосфатов, а также щелочность и растворимую кремнекислоту. В нерастворимой части определяют содержание продуктов коррозии конструкционных материалов и кремнекислоты. Иногда при обработке навески отложений водой получается мутный фильтрат, содержащий, по-видимому, коллоидную кремнекислоту, что затрудняет определение содержания хлоридов, сульфатов и других компонентов. В таких случаях следует отказаться от разделения на растворимую и нерастворимую части, а выполнять анализ из одной навески, определяя в ней все компоненты, кроме хлоридов. Потерю при прокаливании для отложений из проточной части паровых турбин не определяют.
2.4.1 Разложение материала и определение содержания кремнекислоты
Навеску отложений от 0,5 до 0,8 г, взвешенную на аналитических весах, помещают в химический стакан или фарфоровую чашку, слегка смачивают дистиллированной водой и закрывают часовым стеклом. Затем, сдвинув стекло, осторожно вливают в нее 10 - 15 см3 концентрированной соляной кислоты. После окончания выделения газов, свидетельствующих о присутствии в отложениях карбонатов, часовое стекло обмывают в чашку или стакан и выпаривают жидкость на водяной бане или слабо нагревающей плитке почти досуха. При этом стеклянной палочкой растирают обрабатываемый материал, не допуская его цементации. Затем вновь приливают к почти сухому осадку 10 см3 соляной кислоты и опять выпаривают почти досуха. Стеклянной палочкой тщательно растирают материал, добиваясь полного его размельчения. В заключение обрабатывают материал 20 - 30 см3 горячей дистиллированной воды, подкисляют 2 - 3 см3 концентрированной соляной кислоты и отфильтровывают не растворившийся осадок на плотный беззольный фильтр, собирая фильтрат в мерную колбу вместимостью 250 или 500 см3. Осадок на фильтре промывают горячей подкисленной соляной кислотой, дистиллированной водой, собирая промывные воды в ту же колбу. Промывание ведут до исчезновения в фильтрате положительной реакции на присутствие железа (проба с сульфосалициловой кислотой: несколько капель фильтрата смешивают с раствором, содержащим 2 % сульфосалициловой кислоты и 1 % персульфата аммония. Для пробы достаточно 1 - 2 см3 такого раствора. Отсутствие розовой окраски свидетельствует об окончании промывания осадка). Если отложения плохо растворяются в соляной кислоте, то их переводят в растворимое состояние, обрабатывая царской водкой или сплавляя с содой или щелочью.
Промытый осадок помещают в платиновый тигель, сжигают фильтр, осадок прокаливают. Если осадок после прокаливания лишь слабо окрашен, то считать его только кремнекислотой. В таком случае и само прокаливание можно выполнять в фарфоровом, а не в платиновом тигле. Если же осадок явно содержит кроме кремнекислоты оксиды металла, то в платиновый тигель вливают 2 - 5 см3 плавиковой (фтористо-водородной) кислоты и две-три капли серной кислоты, жидкость выпаривают на плитке, прокаливают остаток в муфеле и взвешивают. Потеря веса материала и относится к кремнекислоте; ее выражают в процентах к навеске материала. Остаток после обработки не растворившегося в соляной кислоте материала плавиковой кислотой обычно состоит из оксидов железа. Его относят к содержанию железа. Обычно количество этого остатка весьма невелико. Фильтрат в мерной колбе после доведения объема жидкости до метки тщательно перемешивают. Им пользуются для определения содержания прочих компонентов отложений.
Массовую долю кремнекислоты (%), если она не содержала оксидов металлов, вычисляют по формуле
![]() (1)
(1)
и, если применялась плавиковая кислота для удаления SiF4, по формуле
![]() (2)
(2)
где А0 - вес пустого прокаленного тигля, г;
А1 - вес тигля с осадком кремнекислоты, г;
А2 - вес тигля с остатком после удаления кислот HF и H2SO4 выпариванием и прокаливания тигля с остатком, г;
G - навеска отложений, г.
2.4.2 Определение массовой доли железа
Из мерной колбы, содержащей фильтрат после отделения кремнекислоты, отбирают пипеткой от 10 до 100 см3 жидкости в зависимости от предполагаемого содержания железа в анализируемом материале. Отобранную порцию помещают в коническую колбу, всыпают в нее около 0,2 г персульфата аммония, нагревают до кипения, доливают горячей дистиллированной водой до общего объема примерно 100 см3, приливают 0,5 см3 раствора сульфосалициловой кислоты с массовой концентрацией 0,3 г/см3, нагревая до 60 - 70 °С, и титруют окрашенную в красный или темно-вишневый цвет жидкость 0,05 м раствором трилона, прибавляя титрант в конце титрования по каплям и тщательно перемешивая жидкость. Окончанием титрования является изменение окраски жидкости на желтовато-зеленую. В присутствии значительных количеств цинка или меди титрование протекает нечетко. В таком случае необходимо отделить железо от меди и цинка. Для этого отобранную порцию раствора из колбы, содержащей фильтрат после отделения кремнекислоты, помещают в химический стакан, прибавляют примерно 0,2 г персульфата аммония и осаждают железо аммиаком, приливая его концентрированный раствор до появления слабого, но отчетливого запаха. Осадок гидрооксида железа отфильтровывают на быстрофильтрующий фильтр, промывают 2 - 3 раза горячей дистиллированной водой и фильтр вместе с осадком переносят в тот же стакан, где выполнялось осаждение железа. Вливают в этот стакан 100 см3 горячего раствора соляной кислоты (2 - 3 см3 концентрированной кислоты на 100 см3 дистиллированной воды) и перемешивают стеклянной палочкой до полного растворения гидрооксида железа. После полного растворения осадка приливают 0,5 см3 раствора сульфосалициловой кислоты с массовой концентрацией 0,3 г/см3 и титруют окрашенную в красный или вишневый цвет жидкость 0,05 м раствором трилона до изменения окраски на желто-зеленую. Массовую долю железа (%) в пересчете на оксид трехвалентного железа вычисляют по формуле
![]() (3)
(3)
где А - расход 0,05 м раствора трилона, см3;
М - номинальная молярность раствора трилона (в данном случае М = 0,05);
К - поправочный коэффициент к номинальной молярности раствора трилона;
В - объем мерной колбы, в которую собран фильтрат после отделения кремнекислоты, см3;
П - объект пипетки, т.е. аликвотной порции, отобранной для определения железа, см3;
G - навеска, г;
79,46 - половина молекулярной массы оксида трехвалентного железа;
100 в числителе и 1000 в знаменателе - перерасчет в процентах и перевод граммов в миллиграммы.
Осадок гидрооксида железа содержит также гидрооксид алюминия, который остается в нем и после переосаждения. При желании содержание алюминия может быть определено прокаливанием и взвешиванием переосажденного осадка гидрооксидов. Массовую долю алюминия (%) вычисляют по разности
(Al2О3) = (R2О3) - (Fe2О3).
Сумму массовых долей полуторных оксидов R2О3 (%) получают по формуле
![]() (4)
(4)
где а - вес осадка прокаленных полуторных оксидов, т.е. Fe2О3 + Аl2О3, г.
2.4.3 Определение массовой доли меди
Аммиачный фильтрат после осаждения железа может быть использован для определения содержания меди. В этом фильтрате медь находится в виде окрашенного в интенсивно-синий (точнее в сине-фиолетовый) цвет аммиачного комплекса. Однако при осаждении гидрооксида железа некоторая часть меди соосаждается и, следовательно, задерживается в осадке гидрооксида железа. Для более точного определения содержания меди делается переосаждение: осадок гидрооксида железа после его отделения от жидкости растворяют в небольшом количестве соляной кислоты. Растворение проводят на фильтре, собирая раствор в тот же стакан, в котором выполнялось осаждение гидрооксида железа. Промыв фильтр горячей водой (промывные воды собирают в тот же стакан), нагревают эту жидкость и вновь осаждают железо аммиаком. Осадок отфильтровывают, собирая фильтрат вместе с первым. Теперь потери меди с осадком гидрооксида железа существенно уменьшены. Общий фильтрат упаривают до объема 50 - 60 см3 и далее используют для определения содержания меди.
Для отделения железа от меди может быть использован также следующий способ: к отобранной порции раствора прибавляют примерно 3 г хлористого аммония, растворяют его, охлаждают жидкость до 0 - 5 °С и осаждают железо аммиаком, добавляя его до рН 9,5 - 9,8 (тимолфталеин должен окрашиваться в синий цвет). Необходимое количество аммиака для доведения рН до 9,5 - 9,8 определяют в параллельной пробе жидкости, к которой не прибавляют хлористый аммоний и которую не охлаждают. Осадок отфильтровывают, на фильтре промывают небольшим количеством (до 20 см3) нагретого раствора аммиака с массовой концентрацией 0,05 г/см3. После отделения меди от железа в растворе, содержащим медь, будут присутствовать цинк, кальций, магний. Если содержание меди значительно, на что указывает синяя окраска аммиачного раствора, то его с достаточной точностью можно определить колориметрически по интенсивности этой окраски. Для построения градуировочного графика готовят раствор, содержащий 1 г СuО в 1 см3 (3,138 CuSО4 × 5H2О растворяют в мерной литровой колбе в дистиллированной воде, доводят объем до метки и хорошо перемешивают). К различным порциям этого раствора, помещенным в мерные колбы вместимостью по 100 см3, приливают по 10 см3 концентрированного раствора аммиака с массовой концентрацией 0,25 г/см3, доливают дистиллированной водой до метки, перемешивают и измеряют интенсивность окрасок на фотоколориметре КФК в кюветах длиной 100 мм со светофильтром l = 750 нм. В качестве раствора сравнения служит дистиллированная вода. По результатам колориметрирования строят градуировочный график, откладывая по оси абсцисс содержание меди в пробах, т.е. в мерных колбах вместимостью по 100 см3, а по оси ординат - показания фотоколориметра. Полученные точки соединяют линией. Пользуясь этим графиком, определяют содержание меди. Для этого фильтраты после отделения железа собирают в мерную колбу вместимостью 100 см3, доводят до метки, перемешивают и измеряют интенсивность окраски в кюветах длиной 100 см3 со светофильтром l = 750 нм; раствором сравнения служит дистиллированная вода.
В тех случаях, когда фильтрат, содержащий медь, не вмещается в мерную колбу емкостью 100 см3, его предварительно упаривают до конечного объема 50 - 60 см3. В процессе упаривания часто происходит помутнение жидкости. Тогда к ней приливают 2 - 3 см3 концентрированной соляной кислоты, растворяющей возникшую муть, жидкость переносят в мерную колбу вместимостью 100 см3, приливают в нее 10 - 15 см3 раствора аммиака с массовой концентрацией 0,25 г/см3, дистиллированной водой доливают до метки, тщательно перемешивают и измеряют интенсивность образовавшейся окраски на фотоколориметре.
Массовую долю меди (%) вычисляют по формуле
![]() (5)
(5)
где А - результат колориметрирования, т.е. массовая доля меди в соединенном фильтрате, мг.
Определить массовые доли меди и цинка можно трилонометрическим методом. В коническую колбу отбирают пипеткой 10 - 20 см3 раствора после отделения кремнекислоты, разбавляют очищенной водой до 100 см3 и добавлением аммиака 1:1 доводят до рН ~ 3 по индикаторной бумаге. Добавляют 10 см3 раствора двухзамещенного лимонно-кислого аммония с массовой концентрацией 20 %, 1 - 2 см3 трилоната меди (смешивают точно эквивалентные количества 0,1 н растворов сульфата меди и трилона Б), 5 - 7 капель спиртового раствора индикатора ПАН с массовой долей 0,1 % и медленно при интенсивном перемешивании титруют 0,05 м раствором трилона Б до изменения окраски из фиолетово-красной на желтую. Так как медь и цинк имеют близкие по значению атомные массы, их общую массовую долю (%) в пересчете на оксиды вычисляют по формуле
![]() (5а)
(5а)
где A1 - расход титранта, см3;
К - поправочный коэффициент к данной молярности;
0,05 - молярность раствора трилона Б;
Vисх. - общий объем исходного раствора отложений, см3;
Vпр. - объем исходного раствора отложений, взятый для анализа, см3;
В - масса навески отложений, г;
80 - молекулярная масса (усредненная) CuO + ZnO;
100 в числителе и 1000 в знаменателе - проценты и пересчет навески в миллиграммы.
2.4.4 Определение массовой доли цинка
При щелочной обработке порции фильтрата после отделения кремнекислоты осаждают железо, медь и магний; в растворе остаются цинкаты, алюминаты и кальций. Содержание последнего в отложениях обычно незначительно, и в полученном щелочном растворе цинк может быть определен.
Определение выполняют следующим образом: отобранную пипеткой порцию раствора после отделения кремнекислоты помещают в химический стакан, нагревают и к ней приливают 10 - 15 см3 раствора едкого натра с массовой концентрацией 0,1 г/см3, не содержащего карбонатов. Образующийся осадок отделяют фильтрованием, промывают на фильтре горячей дистиллированной водой, фильтрат и промывные воды собирают в мерную колбу вместимостью 250 см3. Объем жидкости в колбе доливают до метки дистиллированной водой и хорошо перемешивают. Отбирают пипеткой 100 см3 раствора в коническую колбу, всыпают в нее 1 г хлористого аммония, перемешивают до полного растворения соли, прибавляют несколько капель индикатора и титруют 0,05 м раствором трилона Б до изменения окраски индикатора. В качестве индикаторов пригодны: эриохром черный Т (кислотный хром черный специальный ЕТ 00) - переход окраски от винно-красной к синей; кислотный хром синий К - переход окраски от розовой к серо-голубой; пирокатехиновый фиолетовый - переход окраски от зеленовато-голубой к красно-фиолетовой; эриохром сине-черный - переход окраски от красной к синей. Массовую долю (%) в пересчете на оксид вычисляют по формуле
![]() (6)
(6)
где A1 - расход титранта, см3;
81,37 - молекулярная масса оксида цинка;
250 в числителе и 100 в знаменателе - объемы колбы, в которую был собран щелочной раствор цинка, и пипетки, которой была отобрана порция щелочного раствора для титрования.
Следует отметить, что в отобранной порции щелочного раствора присутствует не только цинк, но и кальций и алюминий. Установлено, что алюминий не мешает титрованию цинка, а кальций титруется вместе с ним. Таким образом, расход титранта A1 включает также и расход «на кальций». Обычно в анализируемых материалах содержание кальция незначительно, если же считают необходимым ввести поправку на содержание кальция, то проводят его определение. Возможно определение суммы массовых долей меди и цинка трилонометрическим методом по п. 3.6.5.
2.4.5 Определение массовой доли кальция
Из колбы на 250 см3, куда собран щелочной раствор, отбирают пипеткой 50 см3 раствора и помещают в коническую колбу. Затем вводят 2 см3 свежеприготовленного раствора сульфида натрия с массовой концентрацией 0,1 г/см3 и несколько капель того же индикатора, который применялся при титровании цинка. После этого титруют кальций 0,05 м раствором трилона Б до изменения окраски индикатора. Если после прибавления сульфида натрия жидкость окрасилась в темный цвет, то перед титрованием ее профильтровывают через быстрофильтрующий фильтр, который затем 1 раз промывают небольшим количеством дистиллированной воды. Массовую долю оксида кальция (%) вычисляют по формуле
![]() (7)
(7)
где А2 -расход титранта, см3;
56,08 - молекулярная масса оксида кальция;
250 в числителе и 50 в знаменателе - объемы колбы, в которую собран щелочной фильтрат, и пипетки, которой отобрана аликвотная часть щелочного раствора для титрования кальция, см3.
Для уточнения содержания цинка следует из расхода титранта А1 вычесть удвоенный расход титранта А2, т.е. в формулу для расчета содержания цинка вместо А1 ввести разность А1 - 2А2.
2.4.6 Определение суммы массовых долей кальция и магния
Отбирают пипеткой порцию раствора после отделения кремнекислоты, помещают ее в химический стакан, нагревают до начала кипения и осаждают все металлы, кроме кальция и магния (а также натрия), аммиаком и сульфидом натрия. Сначала в горячий раствор вливают (под вытяжным шкафом) 5 см3 раствора сульфида натрия с массовой концентрацией 0,1 г/см3. При этом происходит выделение сероводорода и осаждение меди и цинка в виде сульфидов, а также части железа. Затем в жидкость вводят избыток аммиака (5 см3 концентрированного раствора аммиака) и продолжают нагревать, образовавшийся осадок отфильтровывают на быстрофильтрующий фильтр, промывают несколько раз дистиллированной водой, собирая фильтрат и промывные воды в коническую колбу. К собранной жидкости прибавляют несколько капель индикатора хром темно-синего или хромоген черного и титруют жидкость 0,05 м раствором трилона Б до изменения цвета индикатора от винно-красного до синего. Сумму массовых долей кальция и магния условно выражают в процентах СаО по формуле
![]() (8)
(8)
где А - расход титранта, см3;
56,08 - молекулярная масса СаО.
Массовая доля магния в отложениях (%) может быть вычислена по формуле
![]() (9)
(9)
Содержание всех этих компонентов в отложениях обычно невелико, их можно определять и на атомно-абсорбционном спектрофотометре. Возможно определение суммы массовых долей кальция и магния по п. 3.6.9.
2.4.7 Определение массовой доли сульфатов
Аликвотную часть раствора из колбы после отделения кремнекислоты и нерастворимых в соляной кислоте веществ помещают в химический стакан, нагревают до слабого кипения и постепенно малыми порциями, а сначала по каплям, добавляют раствор хлористого бария с массовой концентрацией 0,05 г/см3. (Если содержание сульфатов значительно, то жидкость мутнеет, в ней появляются кристаллы сульфата бария. Общее количество хлористого бария не должно превышать 5 см3). Жидкость продолжают слабо нагревать, пока осадок не соберется на дне стакана. Тогда приливают еще около 1 см3 раствора хлористого бария, отмечая - появится муть или нет. В случае появления мути добавляют еще 5 см3 хлористого бария, хорошо перемешивают жидкость и оставляют на сутки. Затем осадок отфильтровывают на плотный беззольный фильтр, промывают на фильтре дистиллированной водой до исчезновения в фильтрате ионов хлора (проба с раствором азотнокислого серебра), переносят во взвешенный прокаленный тигель, подсушивают, озоляют и прокаливают до полного сгорания углистых частичек. Вес полученного сернокислого бария пересчитывают на SO3 и переводят в проценты по формуле
где А - масса сернокислого бария, г;
233,4 и 80,06 - соответственно молекулярные массы сернокислого бария и сульфата в пересчете на SO3.
2.4.8 Определение массовой доли фосфатов
Для определения фосфатов применяют колориметрический способ, переводя фосфорную кислоту в гетерокомплексную фосфорно-молибденовую, окрашенную в желтый цвет, или восстанавливая эту гетерокислоту до синего соединения хлористым оловом. Результаты получают, пользуясь градуировочным графиком, предварительно построенным по стандартным растворам фосфорно-натриевой или фосфорно-калиевой соли. Для выполнения анализа готовят следующие реактивы:
1) раствор молибденово-кислого аммония (5 г этой соли растворяют в 100 см3 дистиллированной воды);
2) раствор хлористого олова - 2,5 г хлорида олова SnCl2 × 2Н2O растворяют в 100 см3 чистого глицерина в фарфоровой чашке при нагревании на кипящей водяной бане. Раствор устойчив против окисления. Хранить его следует в стеклянном сосуде;
3) стандартный раствор фосфата натрия (1 г динатрийфосфата Na2HPO4 × 12Н2O растворяют в 1 л дистиллированной воды).
Титр раствора устанавливают, отбирая порции по 100 см3 в конические колбы и титруя их раствором кислоты или щелочи концентрации с (НСl, NaOH) = 0,1 моль/дм3 (0,1 н) в присутствии индикаторов метилоранжа и фенолфталеина. Раствор титруют сначала в присутствии метилоранжа до изменения окраски. Затем жидкость нагревают до кипения, кипятят около минуты, быстро охлаждают, прибавляют 5 - 7 капель раствора фенолфталеина и титруют раствором щелочи до появления устойчивой бледно-розовой окраски, не исчезающей в течение минуты. Переходной окраски по метилоранжу добиваются, применяя раствор кислоты или щелочи. Проводят три - четыре таких титрования и вычисляют среднее арифметическое. Следует заметить, что расхождения результатов не должны превышать 0,1 см3. Концентрацию РO4, мг/см3, в приготовленном растворе получают по формуле
![]() (11)
(11)
где А - расход раствора щелочи концентрации с (NaOH) = 0,1 моль/дм3 (0,1 н) на титрование 100 см3 раствора динатрийфосфата, см3;
94,97 - молекулярный вес РO4;
К - поправочный коэффициент к децинормальной концентрации для расчета щелочи.
Приготовленный стандартный раствор устойчив и подлежит хранению в склянке с пришлифованной пробкой. Разбавлением этого раствора готовят стандартный раствор, содержащий 10 мг РО4 в 1 дм3. Для этого отмеряют пипетками объем, равный V, см3, стандартного раствора, в мерную колбу вместимостью 1 дм3, доливают дистиллированной водой до метки и перемешивают.
Объем V определяют по формуле
![]() (12)
(12)
Например (РO4) = 0,260 мг/см3, тогда
![]() (13)
(13)
Различные объемы приготовленного раствора динатрийфосфата от 5 до 40 см3 помещают в мерные колбы вместимостью по 50 см3, вводят в каждую по 5 см3 раствора серной кислоты с массовой концентрацией 0,1 г/см3 и по 2 см3 раствора молибденово-кислого аммония. Объемы растворов в колбах доливают почти до метки дистиллированной водой. Спустя 5 мин. добавляют по 0,5 см3 раствора хлористого олова и хорошо перемешивают. Через 3 мин. измеряют интенсивность окрасок со светофильтрами l = 750 нм на фотоколориметре КФК, пользуясь кюветами длиной 50 мм. Раствором сравнения служит дистиллированная вода. Показания фотоколориметра откладывают по оси ординат, а соответствующие им содержания РO4 в пробах - по оси абсцисс, точки соединяют прямой линией.
Для определения содержания фосфатов некоторый объем раствора после отделения кремнекислоты помещают в мерную колбу вместимостью 50 см3, вводят в нее 5 см3 раствора серной кислоты с массовой концентрацией 0,1 г/см3 и 2 см3 молибдатного раствора, доливают дистиллированной водой почти до метки, перемешивают и через 5 мин. добавляют 0,5 см3 раствора хлористого олова. Хорошо перемешав раствор, через 3 мин. измеряют интенсивность окраски. Измерение ведут со светофильтром l = 750 нм, используя в качестве раствора сравнения такой же объем анализируемого раствора, разбавленный в колбе на 50 см3 дистиллированной водой. Массовую долю фосфатов в пересчете на РO4 выражают в процентах к навеске по формуле
![]() (14)
(14)
где А - содержание РO4, полученное по градуировочному графику, мг.
2.5 Анализ водорастворимой вытяжки
Анализ водорастворимой части отложений выполняют следующим образом: навеску отложений помещают в химический стакан, вливают в него 200 - 250 см3 горячей дистиллированной воды, стеклянной палочкой размешивают навеску и нагревают жидкость до слабого кипения, которое поддерживают около часа. Затем нерастворившийся остаток отфильтровывают на беззольный фильтр, собирая фильтрат и промывные воды в мерную колбу вместимостью 500 см3. Промывание остатка ведут горячей дистиллированной водой. Если фильтрование проходит очень медленно, а фильтрат получается мутным, то прекращают фильтрование, берут другую навеску и повторяют всю процедуру ее обработки горячей дистиллированной водой.
Затем, не применяя фильтрования, всю жидкость вместе с нерастворившимся остатком переносят в мерную колбу вместимостью 500 см3, смывая в нее по возможности весь осадок горячей дистиллированной водой. После полного остывания жидкости доводят ее объем до метки дистиллированной водой, тщательно и многократно перемешивают и оставляют отстаиваться.
Обычно через 2 - 3 суток жидкость над осадком осветляется, после чего можно начинать анализ водорастворимой части. Анализ нерастворившегося материала выполнить при этом нельзя. Для установления его состава делают анализ всех материалов без разделения на растворимую и нерастворимую части, как описано выше. Для определения состава водорастворимой части прозрачную отстоявшуюся жидкость осторожно отбирают пипеткой и определяют ее щелочность, содержание в ней хлоридов, сульфатов, кремнекислоты, суммарное содержание щелочно-земельных металлов, т.е. кальция и магния.
Содержание железа, меди, цинка и общее содержание кремнекислоты определять в водной вытяжке не следует, так как водный раствор обычно имеет щелочную реакцию и, следовательно, в раствор перечисленные примеси перейти не могут.
2.5.1 Определение щелочности
Отобранную пипеткой порцию отстоявшейся осветленной жидкости титруют раствором соляной кислоты концентрации с (НСl) = 0,1 моль/дм3 (0,1 н) с индикаторами метилоранжем и фенолфталеином. В отобранную порцию жидкости, помещенную в коническую колбу, вводят 5 - 7 капель раствора фенолфталеина и, если жидкость окрасилась в розовый или красный цвет, титруют ее раствором кислоты до обесцвечивания. Расход раствора кислоты концентрации с (НСl) = 0,1 моль/дм3 (0,1 н) обозначают Аф.ф, см3. После этого вводят 3 - 4 капли раствора метилоранжа и продолжают титрование раствором кислоты до изменения цвета жидкости от желтого до оранжевого (не красного!). Общий расход кислоты, т.е. с самого начала титрования, обозначают Ам.ор., см3. После этого добавляют в избыток еще 2 - 3 см3 раствора кислоты. Жидкость приобретает красный цвет, ее кипятят 1 - 2 мин., по возможности быстро охлаждают и раствором щелочи концентрации с (NaOH) = 0,1 моль/дм3 (0,1 н) доводят окраску до оранжево-желтой. Не отмечая расход щелочного раствора на эту операцию, продолжают титрование до появления неисчезающей окраски индикатора фенолфталеина. Расход щелочи на это титрование обозначают Аобр., см3. По этим данным проводят расчет щелочности, пересчитывая ее на содержание едкого натра, соды и фосфата натрия. Расчеты эти условны, так как, например, едкий натр присутствовать в отложениях не может вследствие высокой растворимости его в паре. Мнимое присутствие едкого натра в отложениях обусловлено образованием сложных соединений, подвергающихся гидролитическому разложению при водной обработке с переходом в раствор едкого натра. При обратном титровании происходит нейтрализация второго Н+ иона фосфорной кислоты, образовавшейся при нейтрализации жидкости по метилоранжу. Но, кроме фосфорной кислоты, расход щелочного раствора в присутствии фенолфталеина может быть обусловлен примесями различных органических кислот, также частично освобождаемых при нейтрализации жидкости по метилоранжу. Все это указывает на условность расчетов компонентов щелочности
![]() (15)
(15)
![]() (16)
(16)
![]() (17)
(17)
где NaOH, Na2CO3 и РO4 - их содержания в отложениях, %;
А (с соответствующими индексами) - расходы кислоты и щелочи растворов концентрации с (НСl, NaOH) = 0,1 моль/дм3 (0,1 н) на титрования, см3;
Кк, Кщ - поправочные коэффициенты к децинормальным растворам кислоты и щелочи;
4; 5,3 и 9,497 - эквиваленты для едкого натра, соды и РO4, умноженные на 0,1.
2.5.2 Определение массовой доли хлоридов
Порцию осветлившегося раствора, отобранную пипеткой, помещают в коническую колбу, приливают 2 см3 концентрированной азотной кислоты и дистиллированной воды до общего объема примерно 100 см3. Затем вводят на кончике ножа несколько кристаллов нитропруссида натрия и три капли раствора метилоранжа. Кристаллы нитропруссида натрия растворяют и титруют окрашенную в оранжево-красный цвет жидкость раствором нитрата ртути II такой концентрации, чтобы каждый миллилитр раствора отвечал 1 мг хлор-иона. Титрование ведут до появления мути в жидкости, которая хорошо заметна при прибавлении одной капли ртутного раствора на фоне черной глянцевой бумаги. Массовую долю хлоридов (%) в пересчете на хлористый натрий получают по формуле
![]() (18)
(18)
где А - расход титранта, т.е. раствора азотнокислой ртути, см3;
Т - содержание хлор-ионов, которому соответствует 1 см3 ртутного раствора, мг/см3;
58,443 и 35,453 - молекулярные веса хлористого натрия и хлор-иона.
2.5.3 Определение массовой доли сульфатов
Порцию прозрачного раствора, отобранную пипеткой из колбы, помещают в химический стакан, приливают 2 см3 концентрированной соляной кислоты и нагревают до кипения. Затем в раствор приливают малыми порциями, а вначале по каплям раствор хлористого бария с массовой концентрацией 0,05 г/см3. Образующийся осадок сернокислого бария после суточного выстаивания отфильтровывают на плотный беззольный фильтр, промывают дистиллированной водой до исчезновения в промывных водах хлоридов (проба с раствором азотнокислого серебра), переносят во взвешенный фарфоровый тигель, подсушивают, озоляют и прокаливают до полного сгорания углистых частиц. После охлаждения тигель с осадком сульфата бария взвешивают. Вес сернокислого бария пересчитывают на SO3 и выражают в процентах к навеске [ф-ла (10)].
2.5.4 Определение суммы массовых долей кальция и магния
К порции осветлившейся жидкости, отобранной пипеткой и помещенной в коническую колбу, добавляют 5 см3 буферной аммиачной смеси и три - четыре капли индикатора хром темно-синего. После этого окрашенный в винно-красный цвет раствор титруют 0,05 м раствором трилона до изменения окраски на синюю. Следует заметить, что даже следы ионов меди или других металлов препятствуют титрованию, затрудняя изменение окраски жидкости. В таком случае перед титрованием к жидкости прибавляют несколько кристаллов диэтилдитиокарбамината натрия. Он связывает ионы, препятствующие титрованию, и процесс происходит нормально. Расход трилона Б пересчитывают на СаО и выражают в процентах к навеске. Для расчета служит формула
![]() (18)
(18)
где А - расход трилона на титрование пробы, см3;
К - поправочный коэффициент к нормальной молярности раствора трилона;
56,08 - молекулярная масса СаО.
2.5.5 Оформление результатов
Результаты осмотра оборудования, определение количества и химического состава отложений заносят в журнал осмотра теплосилового оборудования. Данные анализов отложений с каждой ступени выражают процентным содержанием кремниевой кислоты, оксидов (железа, меди, натрия, кальция, магния, цинка и т.д.), щелочи, а также вес всех этих компонентов на данной ступени.
3 МЕТОДИКА КОНТРОЛЯ СОСТОЯНИЯ КОТЕЛЬНЫХ ТРУБ И ОПРЕДЕЛЕНИЯ КОЛИЧЕСТВА ОТЛОЖЕНИЙ
3.1 Организация контроля
Наиболее доступным и представительным способом контроля за состоянием внутренней поверхности экранных труб является периодическая вырезка контрольных образцов в зоне максимального теплового потока. В отдельных случаях, в основном при пуске головных котлов и при исследовании причин повреждения экранных труб, а также в случае использования одного жидкого топлива или газа с добавкой более 50 % мазута, вместе с этим применяется наблюдение за температурой металла труб с помощью установки специальных термометрических вставок.
На рисунке 1 показано устройство такой вставки для гладких экранных труб, на рисунке 2 - устройство и установка вставки для экранов с плавниковыми трубами, на рисунке 3 - для топочных экранов барабанных котлов.
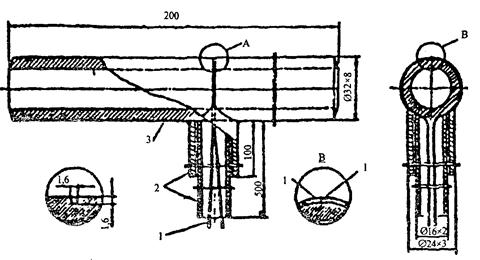
Рисунок 1 - Устройство температурной вставки для гладких экранных труб прямоточных котлов
Все термоэлектрические преобразователи (термопары) располагают на тыльной стороне вставки, собирают в жгут и выводят наружу через штуцер, на выходе из которого установлена клеммная сборка (см. рисунок 2). Их показания выводят на регистрирующий прибор, периодически включаемый для фиксации температуры в течение 2 - 3 ч.
По каждому котлоагрегату ведут журнал учета установленных температурных вставок (таблица 2). Показания термообразующих преобразователей вставок записывают в специальный журнал регистрации температуры металла экранных труб в зоне обогрева (таблица 3) 1 раз в 5 - 7 суток. После накопления опытных данных возможна запись 1 раз в месяц. Колебания температур металла по вставкам фиксируют и заносят в те же журналы. Показания температурных вставок записывают при номинальной нагрузке и установившемся режиме работы котла. Для записи в журнал выбирается максимальное значение температуры, определяемой в течение 5 - 10 мин. непрерывных записей по каждой вставке.
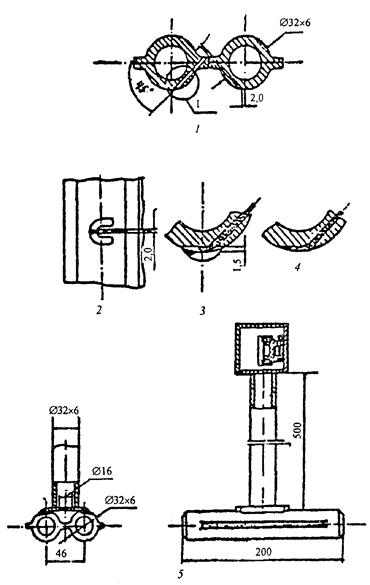
1 - заделка термоэлектрического преобразователя; 2, 3, 4 - заделка термоэлектрического преобразователя по лобовой образующей трубы; 5 - вставка в собранном виде.
Рисунок 2 - Устройство и установка вставки для экранов с плавниковыми трубами прямоточных котлов
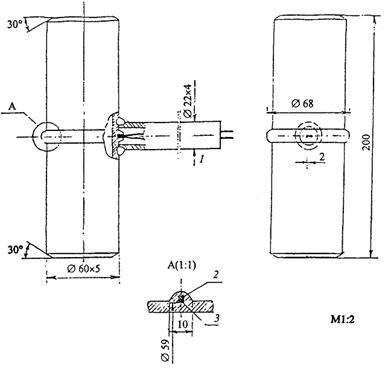
1 - корольки термоэлектрических преобразователей на лобовой образующей трубы; 2 - полоса 2´2; 3 - термоэлектрический преобразователь (термопара).
Рисунок 3 - Температурная вставка для топочных экранов барабанных котлов (р = 16 МПа)
Таблица 2
Журнал учета установленных температурных вставок на котле № ___
типа ________ ГРЭС (ТЭЦ)
|
Номер чертежа изготовления вставки |
Дата установки вставки |
Номер трубы со вставкой |
Экран, на котором установлена вставка |
Номер точки на приборе |
Продолжительность работы вставки*, ч |
Примечание |
||
|
Термопара № 1 |
Термопара № 2 |
|||||||
|
|
|
|
|
|
|
|
|
|
|
* Указывается количество часов работы вставки от момента ее установки до выхода из строя. |
||||||||
Таблица 3
Журнал регистрации температуры металла экранных труб в зоне обогрева котла типа ______ ст. № _____ ГРЭС (ТЭЦ)
|
Дата измерения |
Нагрузка котла, т/ч |
Номер |
Показания термопар |
Температура металла (по вставкам), °С |
Примечание |
||||
|
трубы |
хода |
вставки |
термопары |
||||||
|
Левый боковой |
|
|
|
|
|
|
|
|
|
|
Задний |
|
|
|
|
|
|
|
|
|
|
Правый боковой |
|
|
|
|
|
|
|
|
|
|
Фронтовой |
|
|
|
|
|
|
|
|
|
Данные о времени, местах установки температурных вставок и вырезки образцов записывают в журнал осмотров оборудования в химическом цехе, в него дополнительно заносят и результаты визуального осмотра внутренних поверхностей разрезанных труб, количество отложений и их химический состав.
Маркировку элементов поверхностей нагрева, где устанавливают температурные вставки или делают вырезки, выполняют следующим образом.
На котлах с вертикально расположенными трубами наблюдатель должен находиться вне топки лицом к фронту котла. Трубы и змеевики нумеруют арабскими цифрами: трубы фронтального и заднего экранов - слева направо по фронту котла, трубы боковых и двухсветных экранов - по направлению от фронта к задней стене.
На прямоточных котлах с горизонтальной навивкой труб маркировку производят снизу вверх с указанием номера змеевика (арабскими цифрами) и номер хода (римскими цифрами).
Установку температурных вставок и вырезку образцов труб производят на поверхностях, расположенных в зонах максимальных теплонапряжений и концентраций примесей, где создаются наиболее благоприятные условия для образования отложений и протекания процессов коррозии. Для прямоточных котлов СКД выбирают зону, отвечающую энтальпии среды 1925 - 2150 кДж/кг.
Зоны труб, из которых необходимо производить вырезки контрольных образцов, а также количество образцов пересматривают в случае существенного изменения конструкции котлоагрегата или режима его эксплуатации (например, при переходе на новый вид топлива, реконструкции горелочных устройств или поверхностей нагрева и т.д.).
3.2 Количество и периодичность вырезок
Места вырезки образцов труб, соответствующие зонам максимальных теплонапряжений, определяются для каждого типа котла и используемого топлива при проведении теплотехнических испытаний головных образцов парогенераторов, уточняются при проведении таких испытаний на серийных образцах.
Вырезку образцов труб проводит персонал котлотурбинного (ремонтного) цеха под общим надзором представителя химического цеха. Представители этих цехов составляют акт о вырезке образцов труб для исследования, указывая в нем: дату установки вырезаемого участка трубы и дату вырезки, номер котла, номер и расположение в котле данных труб, а также результаты внутреннего осмотра барабана и коллекторов котла.
Вырезанные образцы труб должны быть промаркированы представителями вышеуказанных цехов и переданы для выполнения анализов и на ответственное хранение в химический цех.
Периодичность вырезок образцов экранных труб устанавливается районным энергетическим управлением для каждого котла электростанции в зависимости от режима его работы и вида топлива. На котлах, работающих на жидком и газообразном топливе или на их смеси, вырезка образцов экранных труб котлов производится 1 раз в два года. При нарушении водно-химического режима, существенном отклонении качества питательной и котельной воды от норм, росте температуры выше предельной по решению главного инженера вырезки выполняются через более короткие промежутки времени, чем межремонтный период. При реконструкции, связанной с изменением топочного режима (горелки, подача воздуха, рециркуляция газов и т.п.), вырезки образцов производятся через один год. На котлоагрегатах, работающих на твердом топливе и смеси твердого и газообразного, вырезки выполняют не чаще, чем через 15 - 18 тыс. ч. эксплуатации. При существенном отклонении качества питательной воды от норм по решению главного инженера электростанции контроль осуществляют через более короткие промежутки времени.
Вырезку образцов проводят циклично: в первый год вырезают крайнюю трубу из рекомендуемой зоны с наименьшим порядковым номером; затем - трубы с последующими номерами по возрастающей. Следующие вырезки вновь начинают с крайней трубы зоны с наименьшим порядковым номером и цикл повторяют. Для оценки эффективности проведенной химической очистки оборудования контрольные образцы вырезают до и после очистки. До химической очистки вырезают очередные по счету трубы чистого и солевого отсеков или НРЧ, и после химической очистки - рядом стоящие трубы с последующими порядковыми номерами.
В аналогичном порядке вырезают образцы труб из водяных экономайзеров.
Образцы труб вырезают в следующем количестве:
- из водяного экономайзера - не менее, чем по два образца на входе и выходе из I ступени и на выходе из II ступени;
- из экранных труб барабанных котлов - не менее, чем по два образца с каждой стены топочной камеры;
- из чистого отсека фронтового и заднего экранов и солевых отсеков боковых экранов - не менее, чем по два образца;
- из пароперегревателей второй ступени вертикального участка с гибом - не менее, чем по два образца.
На котлах без ступенчатого испарения вырезают также не менее, чем по два образца с каждой стены топочной камеры.
В прямоточных котлах вырезают не менее, чем по два образца с каждой поверхности экрана НРЧ, и, в случае необходимости, также из КПП как высокого, так и низкого давления, затем из СРЧ (входной участок) и НРЧ (выходные участки).
3.3 Определение необходимости и эффективности химической очистки
Длительность межпромывочного периода ограничивается по условию превышения допустимого количества отложений на огневой (лобовой) стороне внутренней поверхности трубы, определяемого по результатам очередной вырезки образцов (таблица 4).
В случае наличия термометрических вставок в экранные трубы упреждающим сигналом о необходимости проведения химической очистки может быть превышение допустимой температуры нагружной поверхности трубы:
- для стали 20 при давлении 15,5 МПа в барабане (труба Æ 60´6 мм), t = 460 °С - по условию длительной прочности (ресурс 100 тыс. ч.);
- для стали 20 при давлении 11,5 МПа и ниже t = 500 °С - по условию жаростойкости (окалинообразования).
При давлении 15,5 МПа температура участков (поясов) экранных труб из стали 15ХМ и 12Х1МФ практически не может быть выше 500 °С - предельной для стали 20 по условию окалинообразования и наружной коррозии (для топлива из сланцев 450 °С).
После эксплуатационной химической очистки вырезаются контрольные образцы для определения оставшегося количества отложений. Для барабанных котлов оно не должно быть больше 70 г/м2, для прямоточных - не более 50 г/м2.
Кроме показаний температурных вставок, при определении необходимости химических очисток котлов можно ориентироваться на предельные значения загрязненности экранных труб, указанные в таблице 4.
Таблица 4
Предельные количества отложений на огневой поверхности экранных труб
|
Топливо, г/м2 |
||||
|
Жидкое и газообразное |
Твердое и жидкое |
Уголь |
Торф, щепа и прочее |
|
|
Барабанные котлы: |
||||
|
давление до 4 МПа |
800 |
800 |
1000 |
1200 |
|
от 4 до 10 МПа |
600 |
600 |
800 |
1000 |
|
от 10 до 15,5 МПа |
400 |
400 |
600 |
800 |
|
Прямоточные котлы: |
||||
|
докритического давления |
300 |
300 |
400 |
- |
|
сверхкритического давления |
200 |
250 |
300 |
- |
|
ГАВР или ГВР |
Для рыхлого слоя |
|||
|
НКВР или КАВР |
70 |
70 |
120 |
- |
|
|
Общая загрязненность |
|||
|
250 |
300 |
400 |
- |
|
При установившемся водно-химическом режиме без четко выраженных его нарушений прогнозирование сроков выхода котла на предельный уровень отложений производят расчетом по усредненным результатам выполненных и предыдущих определений. При значительных расхождениях в результатах анализов по определению количества отложений расчеты производятся по отдельным поверхностям нагрева (фронтовые экраны, задние экраны, экраны 2-й ступени испарения). Порядок расчета следующий:
1) Определяется интенсивность роста отложений G за 1000 ч, г/м2, по формуле
![]() (20)
(20)
где q2 - усредненный показатель только что выполненных определений количества отложений, г/м2;
q1 - усредненный показатель предыдущего определения количества отложений, г/м2;
Dt - время работы котла между настоящей и предыдущей вырезками образцов для анализа.
2) Определяется время работы котла Dt после его пуска для выхода на предельный уровень отложений, ч, по формуле
![]() (21)
(21)
где qп - предельное значение отложений в соответствии с данными таблицы 4.
3.4 Вырезка и подготовка образцов труб для исследования
Согласно заявке химического цеха вырезку образцов выполняют по карте вырезок. Участки труб, расположенные на расстоянии 10 - 15 см от концов вырезанного автогеном образца, не учитывают при определении количества и состава отложений, вследствие перегрева металла, изменения его структуры, характера и состава отложений.
При удалении из котла вырезанные образцы необходимо предохранять от резких ударов и сотрясений, чтобы не нарушить целостность отложений.
На огневой стороне вырезаемого образца наносят метку краской. Затем вырезанные участки труб раскладывают и, по возможности, освобождают их наружную поверхность от сыпучих зольных отложений, не прибегая при этом к ударам и сотрясениям. После этого представитель химического цеха размечает мелом участки длиной 40 - 60 см для описания и снятия отложений и не менее двух участков длиной 5 см в разных местах трубы для определения их загрязненности. Затем на середину каждого намеченного участка трубы с огневой и тыльной сторон наклеивают этикетки со следующими данными: дата вырезки, электростанция, номер котла, номер и расположение трубы в котле, солевой или чистый отсек, огневая или тыльная сторона, стрелкой указан верх или низ трубы.
Вырезанные и промаркированные образцы передают в механическую мастерскую, где их разрезают согласно меловым меткам, нанесенным представителем химического цеха.
После распиливания труб на отдельные участки каждый из них продольно распиливают на огневую и тыльную половины.
Трубы разрезают на фрезерном или продольно-строгальном станке в присутствии представителя химического цеха. При этом должны соблюдаться следующие условия:
- режущий инструмент (фреза, резец) не должен смачиваться (распиливание ведут «всухую»);
- перед пуском станка внутреннюю поверхность трубы нужно осторожно прикрыть толстой бумагой для предотвращения попадания стружки и металлических опилок;
- после распиливания обе половинки трубы необходимо сложить вместе и плотно скрепить.
Представитель химического цеха тщательно осматривает внутреннюю поверхность каждого участка трубы и описывает его состояние, снимает отложения для определения их химического состава и оценивает загрязненность трубы. Все данные заносят в специальный журнал осмотров оборудования.
При описании отложений отмечают их цвет, толщину слоя в различных местах, степень равномерности покрытия поверхности трубы, характерные особенности (пластинки, слоистая или губчатая масса, отдельные бугорки, сплошной слой и т.п.).
В описание включают и характеристику состояний металла под слоем отложений - наличие язвин, их количество, размеры, глубина и характер. Разрушенные экранные трубы необходимо подвергать металлографическому исследованию.
Количественную оценку загрязненности поверхностей нагрева отложениями производят путем вырезки контрольных участков труб, снятия с них отложений и определения их веса и химического состава.
3.5 Снятие отложений
Снятие отложений механическим способом, т.е. с помощью скребков, ножичков и других приспособлений дает возможность при осторожном выполнении получать пробы, пригодные для исследования и химического анализа. Однако достаточно надежную оценку степени загрязненности труб отложениями таким способом получить нельзя, так как он имеет ряд серьезных недостатков: неполнота очистки из-за невозможности удалить отложения из мелких язвин и пор; засорение отложений материалом скребков и металлом трубы при интенсивном снятии накипи; затруднительность снятия прочно скрепленных отложений.
Особо прочно связанные с металлом отложения можно снять путем сминания участка трубы в тисках. Для этого участок трубы длиной 10 см с очищенной наружной поверхностью разрезают вдоль на огневую и тыльную половины, затем одну из них закрепляют в тисках и скребком очищают рыхлые отложения, собирая их в отдельный бюкс или пакет. После этого закрывают отрезок трубы плотным листом бумаги и постепенно сильно сжимают тиски. При сминании металла отложения отскакивают, их пересыпают в другой пакет или бюкс. Обе пробы взвешивают и определяют степень загрязнения ими трубы q, г/м2, по формуле
![]() (22)
(22)
где D - количество снятых рыхлых или плотных отложений, г;
S - измеренная поверхность, с которой сняты отложения, см2.
Достаточно полное снятие отложений достигается также электролитическим способом (см. приложение А).
3.6 Химический анализ отложений
3.6.1 Подготовка отложений к анализу
При эксплуатации паровых котлов на поверхностях нагрева образуются отложения различного состава, что обусловлено как примесями, присутствующими в воде, так и коррозионными процессами. Отложения в экранных трубах паровых котлов, в трубках различных подогревателей и конденсаторов часто называют накипью.
Химический анализ отложений и накипей состоит в определении потери при прокаливании, содержания кремнекислоты, фосфатов, сульфатов, железа, алюминия, меди, цинка, кальция, магния, натрия, а в отдельных случаях и других веществ. В зависимости от типа отложений объем анализа может меняться. Так, в котельных накипях, образовавшихся в трубах барабанных котлов, нет смысла определять содержание растворимых в воде примесей (обычно их нет вследствие контакта с котловой водой). В отложениях из труб котлов прямоточного типа нет смысла определять содержание фосфатов, так как для этих котлов фосфатирование не проводится. Обычно в этих отложениях отсутствуют и органические вещества.
По способам анализа котельные отложения удобно разбить на две группы:
1) отложения, образующиеся в трубах барабанных котлов,
2) отложения, образующиеся в трубах прямоточных котлов и в трубах пароперегревателей.
Учитывая значительное разнообразие составов накипей и отложений, обусловленное различными составами вод, особенностями водно-химических режимов и отличиями эксплуатационных режимов оборудования, ниже предлагаются наиболее простые в осуществлении, но достаточно избирательные и точные способы определения их отдельных составляющих. При комбинировании этих способов, при решении конкретных задач следует учитывать состав накипей и отложений. Их основу составляют соединения (оксиды) железа и цинка, а также металлическая медь и ее оксиды. Повышенное содержание щелочно-земельных металлов указывает на явные дефекты в эксплуатации водоподготовительного оборудования, на присосы охлаждающей воды или на неправильное ведение водно-химических режимов. Поэтому основное внимание в рекомендуемых методиках обращено на определение содержания именно этих компонентов накипей и отложений.
Материал для анализа, т.е. накипь или отложения, нужно подготовить: тщательно измельчить сначала в стальной ступке, а затем в агатовой до состояния пудры (при потирании между пальцами не должно ощущаться крупинок). Перед измельчением должны быть удалены из пробы частички грата, окалины и посторонние предметы (кусочки проволоки, щепки, обрывки материи и пр.).
3.6.2 Определение потери при прокаливании или привеса
Навеску от 0,2 до 0,7 г отбирают во взвешенный, хорошо прокаленный фарфоровый тигель, помещают его в холодный муфель, включают нагревание, прокаливают при температуре 500 - 550 °С в течение 1 ч. Затем охлаждают в эксикаторе, взвешивают и вновь прокаливают при температуре 800 - 850 °С в течение 2 ч. Тигель охлаждают в эксикаторе над прокаленным хлористым кальцием, взвешивают и повторяют прокаливание еще в течение 1 ч. Если при втором взвешивании изменение веса составляет не более 0,7 мг, то определение считается законченным. В противном случае повторяют прокаливание еще в течение часа и вновь взвешивают. Потерю при прокаливании или привес А, %, (если С2 > С1 вычисляют по формуле
![]() (23)
(23)
где С1 - вес тигля с навеской до прокаливания, г;
С2 - вес тигля с навеской после окончательного прокаливания, г.
3.6.3 Определение содержания кремнекислоты
Навеску от 0,5 до 0,8 г отбирают в чистый химический стакан или в чистую фарфоровую чашку с гладкими стенками. Вливают 1 см3 дистиллированной воды, смачивая ею анализируемый материал. Затем добавляют 5 - 7 см3 концентрированной химически чистой соляной кислоты или царской водки, тщательно перемешивают стеклянной палочкой и ставят чашку или стакан на водяную баню, часто перемешивая и растирая образующиеся комочки. После полного испарения кислоты емкость охлаждают и вливают новую порцию кислоты, которую также выпаривают. Так поступают трижды, после чего, смочив сухой остаток несколькими каплями соляной кислоты, вливают 20 - 30 см3 горячей дистиллированной воды, перемешивают и, дав несколько отстояться, отфильтровывают осадок кремнекислоты на плотный беззольный фильтр, собирая фильтрат в мерную колбу вместимостью 250 или 500 см3. Осадок на фильтре промывают горячей дистиллированной водой, подкисленной соляной кислотой (20 см3 кислоты на 500 см3 дистиллированной воды). Промывание ведут до исчезновения желтой окраски осадка и самого фильтра, собирая фильтрат от кремнекислоты и промывные воды в мерную колбу вместимостью 250 и 500 см3. В заключение дважды промывают осадок горячей водой, после чего его вместе с фильтром помещают во взвешенный прокаленный фарфоровый тигель, подсушивают и прокаливают в муфеле при 900 - 950 °С до постоянного веса. Обычно для этого достаточно двух часов.
Если имеются основания предполагать в накипи высокое содержание кремнекислоты, то целесообразно прокаливание осадка вести не в фарфоровом, а в платиновом тигле. В том случае, когда прокаленный осадок окрашен в желто-коричневый цвет, указывающий на его загрязнение оксидами железа, медью и прочими примесями, в платиновой тигель вливают 2 - 3 см3 концентрированной плавиковой кислоты и несколько капель концентрированной серной кислоты. Затем нагреванием удаляют избыток кислот. Вместе с ними улетучивается и кремниевая кислота в виде кремнефторида (SiF6). Тигель прокаливают и взвешивают. Содержание кремнекислоты (%) вычисляют по формулам
В первом случае
![]() (24)
(24)
во втором
![]() (25)
(25)
где C1 - вес тигля с осадком, г;
С0 - вес пустого тигля (фарфорового), г;
С2 - вес платинового тигля после удаления избытка кислот и последующего прокаливания, г.
Если остаток после удаления кремнекислоты значителен (более 5 мг), то его следует растворить в соляной кислоте и раствор прибавить к фильтрату после отделения кремнекислоты. При меньших количествах осадка его следует засчитать как оксиды железа, выразить в процентах к навеске и прибавить к проценту содержания железа, определенного из фильтрата после отделения кремнекислоты.
Некоторые накипи и отложения не полностью разлагаются при обработке их соляной кислотой. Это характерно для недостаточно хорошо измельченных проб. В таких случаях применяют сплавление. Для этого в чистый платиновый тигель всыпают около 0,5 г смеси безводной соды с безводным углекислым калием, на эту смесь помещают навеску анализируемых отложений (накипи), перемешивают смесь платиновой проволочкой и засыпают ее еще некоторым количеством (обычно 0,5 - 0,7 г) смеси углекислых солей калия и натрия. Платиновый тигель со смесью ставят в фарфоровый тигель большего размера и помещают в холодный муфель, температуру в котором постепенно доводят до 850 - 900 °С. При этой температуре тигель выдерживают около часа, затем извлекают из муфеля, дают остыть и погружают в химический стакан, в который затем вливают дистиллированную воду так, чтобы она покрывала лежащий в стакане тигель. Нагрев воду в стакане почти до кипения, добиваются отделения плава от тигля, поворачивая его стеклянной палочкой. Если полностью добиться этого не удалось, покрывают стакан часовым стеклом и очень осторожно, малыми порциями вливают в стакан 25 - 30 см3 концентрированной соляной кислоты. Эту операцию следует выполнять исключительно осторожно, так как при этом происходит вскипание жидкости от выделяющейся углекислоты. Когда вся кислота влита и проверка индикаторной бумажкой подтвердила, что жидкость приобрела кислую реакцию, ее нагревают до слабого кипения, добиваясь полного отделения частиц плава от стенок тигля. Очищенный тигель извлекают из жидкости, обмывают его наружные и внутренние стенки дистиллированной водой, а раствор упаривают почти досуха.
Далее повторяют обработку осадка, описанную выше, т.е. приливают концентрированную соляную кислоту, выпаривают и повторяют эту операцию. Затем отфильтровывают осадок, промывают его, помещают в тигель и прокаливают до постоянного веса. В данном случае кремнекислота всегда чиста, поэтому обработка плавиковой кислотой не нужна. Фильтрат от кремнекислоты и промывные воды собирают в мерную колбу вместимостью 250 или 500 см3. В этом растворе определяют содержание железа, меди и других компонентов отложений. Предварительно объем жидкости в колбе доливают до метки дистиллированной водой и тщательно перемешивают.
3.6.4 Определение массовой доли железа
Из колбы, содержащей фильтрат и промывные воды от кремнекислоты, отбирают пипеткой порцию жидкости, помещают ее в химический стакан, приливают 2 см3 концентрированной азотной кислоты или всыпают около 0,5 г персульфата аммония и нагревают до кипения. Затем, прекратив кипение, вводят по каплям или малыми порциями концентрированный раствор аммиака до полного осаждения гидрооксидов железа и алюминия. Осадок отфильтровывают на беззольный быстрофильтрующий фильтр, промывая и его и фильтр 2 - 3 раза горячей дистиллированной водой. Фильтрат собирают в чистый стакан. Осадок на фильтре растворяют в горячей разбавленной соляной кислоте, промывая затем фильтр горячей водой. Фильтрат и промывные воды собирают в тот стакан, в котором проводилось осаждение гидрооксида железа и алюминия. Частички оксидов, приставшие к стенкам стакана, обмывают дистиллированной водой, жидкость нагревают до 40 - 50 °С и вновь осаждают аммиаком. Осадок отфильтровывают на тот же фильтр, промывая и осадок и фильтр горячей дистиллированной водой. Фильтрат и промывные воды приливают к первому фильтрату. В этом объединенном фильтрате содержатся остальные компоненты накипи.
Осадок гидрооксида железа и алюминия может быть прокален при температуре 900 °С в течение 30 мин. с проверкой постоянства веса и взвешен или растворен, и железо оттитровано раствором трилона. При взвешивании прокаленного осадка гидрооксидов следует иметь в виду, что они содержат кроме Fe2O3 и Аl2O3 фосфаты в виде P2O5. От присутствия фосфатов не освобождает и переосаждение гидрооксидов, основная цель которого - отделение меди и цинка от железа и алюминия. Для определения железа титрованием фильтр с осадком переносят в химический стакан, смачивают 1 см3 концентрированной соляной кислоты, вливают 100 см3 горячей дистиллированной воды, добавляют 0,5 см3 раствора сульфосалициловой кислоты с массовой концентрацией 0,3 г/см3, нагревают жидкость до 70 - 80 °С и титруют горячую жидкость 0,05 м раствором трилона Б до изменения окраски от винно-красной до светло-желтой или почти бесцветной. Массовую долю железа в пересчете на Fe2O3, %, получают по формуле
![]() (26)
(26)
где А - расход титранта, см3;
К - поправочный коэффициент к номинальной молярности раствора трилона Б;
79,46 - половина молекулярного веса Fe2O3;
G - навеска накипи, г;
П - объем пипетки, см3;
В - объем колбы, см3;
100 в числителе и 1000 в знаменателе - пересчет в проценты и перевод миллиграммов в граммы.
3.6.5 Определение суммы массовых долей меди и цинка
Фильтрат от железа содержит медь, цинк, кальций и магний. Для определения суммы меди и цинка фильтрат упаривают примерно до 100 см3, приливают по каплям концентрированную уксусную кислоту до рН от 4 до 5 (значение рН контролируют индикаторной бумажкой), приливают 5 - 7 капель спиртового раствора индикатора ПАН и медленно при интенсивном перемешивании титруют жидкость 0,05 м раствором трилона Б до изменения окраски от фиолетово-красной до желтой. Сумму массовых долей (CuO + ZnO), %, вычисляют по формуле
![]() (27)
(27)
где А - расход титранта, см3;
80 - средняя молекулярная масса оксидов меди и цинка (среднее из 79,55 для оксида меди и 81,37 для оксида цинка).
3.6.6 Определение массовой доли меди
Если аммиачный фильтрат после осаждения железа имеет интенсивную синюю окраску, то это свидетельствует о значительном содержании меди в накипи. В таком случае, повторив осаждение гидрооксида железа, как это описано выше, фильтрат упаривают до объема 15 - 20 см3. Затем прибавлением нескольких капель азотной кислоты растворяют осадок, который может образоваться при выпаривании, количественно переносят жидкость в мерную колбу вместимостью 50 см3 и доливают до метки концентрированным раствором аммиака. Образующийся синий раствор колориметрируют на фотоколориметре, как это описано п. 2.4.3. Там же приведена и формула для вычисления содержания меди.
3.6.7 Определение массовой доли металлической меди
В котельных накипях медь часто присутствует в металлическом состоянии. Для определения ее содержания навеску накипи 0,15 - 0,20 г в конической колбе с пришлифованной стеклянной пробкой обрабатывают 100 см3 насыщенного раствора сернокислого серебра, часто и сильно взбалтывая. Обработку продолжают час, после чего жидкость сливают через бумажный фильтр, стараясь не переносить осадок на фильтр. В колбу вливают еще 25 - 30 см3 насыщенного раствора сернокислого серебра и вновь взбалтывают. Перемешивание и периодическое взбалтывание жидкости с осадком продолжают еще час, после чего профильтровывают через тот же фильтр, собирая оба фильтрата и промывные растворы в мерную колбу вместимостью 250 см3. Промывание осадка и фильтра ведут дистиллированной водой. Доведя объем жидкости в колбе до метки, ее перемешивают и определяют содержание меди колориметрически (см. п. 2.4.3). Массовую долю металлической меди в отложениях и накипях вычисляют (%) по формуле
![]() (28)
(28)
где а - результаты колориметрирования, т.е. содержание меди, определенное в порции, отобранной из колбы 250 см3, мг;
250 - объем колбы, куда собран фильтрат после обработки навески серно-кислым серебром, см3.
Все растворы после определения содержания металлической меди, а также и осадок отложений после его обработки сернокислым серебром, необходимо использовать для выделения серебра. Следует отметить, что общее количество серебра, израсходованное на одно определение, составляет около 0,7 г.
3.6.8 Определение массовой доли цинка
Содержание цинка может быть определено по разности суммы оксидов меди и цинка и содержания меди, пересчитанного на оксид меди.
3.6.9 Определение суммы массовых долей кальция и магния
Отобрав из мерной колбы аликвотную часть раствора, полученного после отделения кремнекислоты, проводят осаждение железа, как описано в п. 3.6.4. Затем фильтрат от железа обрабатывают диэтилдитиокарбаминатом натрия, всыпая в жидкость примерно 0,2 - 0,3 г этого реактива. Жидкость хорошо перемешивают и отфильтровывают образовавшиеся диэтилдитиокарбаминаты меди и цинка. Осадок промывают 2 раза дистиллированной водой, собирая фильтрат и промывные воды в коническую колбу, вводят несколько капель индикатора хром темно-синего и титруют 0,05 м раствором трилона до изменения красного цвета жидкости на сине-фиолетовый.
Сумма массовых долей кальция и магния выражается процентным количеством СаО в накипи по формуле
![]() (29)
(29)
где А - расход титранта, см3;
56,08 - молекулярная масса СаО.
3.6.10 Определение массовой доли фосфатов
Аликвотную часть раствора из колбы, в которую собран фильтрат от кремнекислоты, помещают в химический стакан, добавляют 15 см3 раствора азотнокислого аммония с массовой концентрацией 0,3 г/см3 и 10 см3 азотной кислоты с массовой концентрацией 0,2 г/см3 (258 см3 концентрированной 1,38 азотной кислоты разбавляют до литра дистиллированной водой). Жидкость нагревают до кипения и вливают в нее тонкой струей 15 см3 раствора молибдата аммония с массовой концентрацией 0,03 г/см3. Нагревание прекращают, жидкость перемешивают вращением стакана и оставляют на 2 - 3 часа. Осадок фосфоромолибдата должен иметь чисто-желтый цвет; светлая беловатая окраска свидетельствует о примеси молибденовой кислоты. В этом случае осаждение необходимо повторить, взяв другую пробу или растворив осадок аммиаком (нагрев жидкость и влив в нее 20 см3 азотной кислоты).
Чисто-желтый осадок отфильтровывают на плотный беззольный фильтр, промывают раствором азотнокислого калия с массовой концентрацией 0,05 г/см3 до нейтральной реакции фильтрата по метилоранжу. Осадок вместе с фильтром помещают в тот стакан, где проводилось осаждение, приливают 50 см3 нагретой дистиллированной воды и несколько капель раствора фенолфталеина. Затем в стакан приливают точно отмеренный объем раствора едкого натра концентрации с (NaOH) = 0,1 моль/дм3 (0,1 н), добиваясь появления вполне устойчивой красной окраски индикатора, после чего оттитровывают избыток щелочи раствором кислоты. Содержание фосфатов может быть выражено или на РO4 или на Р2O5 в процентах. В последнем случае пользуются формулой
![]() (30)
(30)
где Ащ и Ак - соответственно количества щелочи и кислоты, израсходованные на растворение осадка и на титрование избытка щелочи кислотой, см3;
Кщ и Кк - соответственно поправочные коэффициенты к децинормальным растворам щелочи и кислоты;
0,1 - номинальная нормальность растворов щелочи и кислоты;
3,086 - эквивалентный вес Р2O5 в данной реакции.
3.6.11 Определение массовой доли сульфатов
Отобрав аликвотную часть раствора из фильтрата после удаления кремнекислоты, помещают его в химический стакан, нагревают до кипения и по каплям приливают 5 см3 раствора хлористого бария с массовой концентрацией 0,05 г/см3. Нагревание прекращают, дают осадку осесть и приливают к прозрачной жидкости еще несколько капель раствора хлористого бария. Добавление проводят осторожно, чтобы не замутить жидкость. Если в прозрачной жидкости не поднимется муть, осаждение считают законченным, в противном случае ее вновь нагревают и добавляют еще 5 см3 раствора хлористого бария. Жидкость с осадком оставляют на сутки, периодически перемешивая ее. Затем осадок отфильтровывают на плотный беззольный фильтр. На фильтре промывают сначала холодной, а под конец горячей дистиллированной водой до исчезновения в промывной воде кислой реакции или хлоридов (проба с AgNO3). Осадок вместе с фильтром помещают во взвешенный фарфоровый тигель, подсушивают, озоляют фильтр и прокаливают осадок при 700 - 800 °С в муфельной печи. После взвешивания повторяют прокаливание, чтобы убедиться в полном удалении углеродистных частиц фильтра. Массовую долю сульфатов (%) вычисляют по формуле
![]() (31)
(31)
где А - вес сернокислого бария, г;
0,343 - коэффициент пересчета веса сернокислого бария на SO3.
Приложение А
(обязательное)
ОПРЕДЕЛЕНИЕ ЗАГРЯЗНЕННОСТИ ТРУБ МЕТОДОМ КАТОДНОЙ ОБРАБОТКИ ПОВЕРХНОСТИ
А.1 Подготовка контрольных образцов
Контрольные образцы труб вырезают автогеном или любым другим способом. Длина контрольного участка должна быть не менее 40 - 60 см. Участки поверхности образца на расстоянии 10 - 15 см от края не обрабатываются, так как их нельзя считать характерными из-за перегрева металла при резке и попадании сварочного грата.
Вырезанный участок трубы на токарном станке обтачивают снаружи на глубину 0,5 - 0,6 мм для удаления наружных загрязнений. Затем разрезают на кольца длиной 5 см. Для анализа и исследования берут 2 - 3 кольца, которые разрезают на огневые и тыльные полукольца, если необходимо исследовать состояние этих поверхностей по отдельности.
Чтобы при всех вышеперечисленных операциях не разрушить отложения на поверхности образцов, отрезки труб и кольца должны хорошо закрепляться на станках во избежание вибраций. Режущие инструменты нельзя смачивать эмульсией. Недопустимы удары по образцам.
Трубы с внутренним диаметром более 5 см можно не обтачивать снаружи.
А.2 Подготовленные таким способом кольца или полукольца протирают снаружи спиртом для обезжиривания поверхности, высушивают в сушильном шкафу при 105 - 110 °С, охлаждают в эксикаторе с прокаленным хлористым кальцием и взвешивают на аналитических весах с точностью до 0,001 г. До начала исследования образцы хранят в указанном эксикаторе.
Во всех случаях необходимо перед электролитическим снятием отложений скребком удалить рыхлый слой, который взвешивают и исследуют отдельно. Считается, что рыхлый слой отложений имеет иную природу и состав, чем плотный, снимаемый катодным электролизом.
А.3 Схема установки с двумя типами электролитических ванн показана на рисунке А.1. В качестве анодов следует применять свинцовые стержни или пластинки, которые могут быть изготовлены из гранулированного или листового свинца, либо из свинцовой стружки сплавлением металла в стеклянной трубке диаметром 4 - 6 мм и длиной 8 - 10 см.
Трубку устанавливают вертикально, нижнее отверстие затыкают асбестовой пробкой, насыпают частички свинца и вставляют конец зачищенной медной проволоки. Затем газовой горелкой нагревают трубку до температуры плавления свинца, кусочки которого добавляют до тех пор, пока их уровень не достигнет 6 - 8 см. После этого трубку охлаждают, стекло разбивают, а полученный свинцовый стержень с вплавленной в него медной проволокой зачищают. Выступающий конец медной проволоки служит для присоединения свинцового электрода к электролитической схеме. Можно в качестве анода в электролитической ванне использовать угольно-графитные стержни, применяемые в электротехнике.
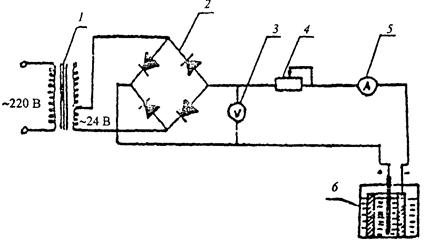
1 - понижающий трансформатор (латр); 2 - выпрямитель с диодами Д24, В; 3 - вольтметр постоянного тока (0 - 30 В); 4 - регулирующий реостат (70 Ом); 5 - амперметр постоянного тока (0 - 03 А); 6 - электролитическая ванна.
Рисунок А.1 - Установка для катодной обработки образцов. (Схема установки)
Рисунок A.2 - Ванны для обработки образцов с внутренним диаметром менее 5 см (а) и более 5 см (б)
А.4 В качестве электролитической ванны используют стакан из полиэтилена, органического или обычного стекла диаметром 6 - 8 см и высотой не менее 5 см.
А.5 Образцы (кольца или полукольца), имеющие внутренний диаметр менее 5 см, обрабатывают в ванне (рисунок А.2, а), погружая их полностью. Поэтому наружная поверхность и торцы колец или полуколец должны быть тщательно очищены от загрязнений. Необходимость защиты наружной поверхности образцов специальными покрытиями зависит от условий проведения электролитической обработки образцов.
А.5.1 Если электролитическая обработка выполняется в растворе моноцитрата аммония с массовой концентрацией 0,1 г/см3 при температуре не выше 70 °С и плотности тока на катоде 1 мА/см2, то при длительности процесса не более 40 мин. заметного растворения металла не происходит. Следовательно, можно не защищать наружную поверхность образцов какими-либо покрытиями. Экспериментально установлено, что в этих условиях достигается удаление отложений, вызванных атмосферной коррозией.
А.5.2 Если электролитическая обработка выполняется в растворе серной кислоты с массовой долей 7 % и уротропина с массовой долей 5 - 7 % и присутствует окалина, раствор серной кислоты берется с массовой долей 10 %, уротропин той же концентрации. В этом случае наружные и торцевые поверхности должны быть защищены.
На практике вполне удовлетворительные результаты дает применение раствора двухзамещенного лимонно-кислого аммония с массовой концентрацией 0,1 г/см3.
А.5.3 При выполнении электролитического снятия отложений в более жестких условиях необходимо защищать наружную и торцевую поверхности образцов специальными покрытиями. Для этого могут быть рекомендованы: эпоксидный клей ЭДП, смешанный с отвердителем; клей БФ-2 или БФ-4; химически стойкие лаки; сплавленная смесь парафина с канифолью в соотношении 1:1.
Покрытия наносят кисточкой на высушенную поверхность, сутки выдерживают на воздухе и досушивают в сушильном шкафу в течение 2 - 3 ч при 80 °С до постоянного веса. При использовании парафино-канифольного покрытия досушка в сушильном шкафу не проводится.
А.6 Для снятия отложений очищенный и взвешенный, а если требуется, то и защищенный специальным покрытием образец 5 помещают в стакан 6 и центрируют вставкой 1 таким образом, что анод 3 находится в центре кольца или полукольца (см. рисунок А.2, а). Фиксированное положение анода по отношению к образцу обеспечивает равномерность очистки и постоянство силы тока по всей его поверхности. В стакан вливают подогретый до 70 - 80 °С электролит 7 так, чтобы края образца выступали над раствором на 5 - 8 мм, после чего стакан закрывают крышкой из органического стекла 2 с укрепленным на ней анодом. Анод должен быть опущен в электролит почти до дна стакана. Через прорезь в крышке выступающий из электролита край кольца или полукольца образца захватывают зажимом типа «крокодил» 4. Причем зажим не должен касаться раствора, иначе будет загрязняться электролит. Сразу же после установки ванны подают напряжение на электроды и при полностью введенном реостате устанавливают заданную величину тока по показаниям амперметра.
А.7 Образцы с внутренним диаметром более 5 см очищают в ванне, изображенной на рисунке А.2, б. Образец ставят в плоскую чашку на слой расплавленного парафина, который после застывания герметично уплотняет дно кольца. Электролит, нагретый до температуры не более 50 °С, вливают внутрь кольца и в его центр опускают анод.
А.8 Травление образцов продолжают 30 мин., после чего их освобождают от электролита, шпателем или палочкой с резиновым наконечником стирают остатки отложений, смывая их струей дистиллированной воды. Визуально определяют, полностью ли произошла очистка образца от отложений. Если обнаруживают остатки («островки») неудаленных отложений, то продолжают процесс, опустив образец в тот же электролит и включив ток. Если же образец полностью освобожден от отложений, его пассивируют, опуская в раствор аммиака с массовой концентрацией 0,05 г/см3, содержащий 0,5 % нитрита натрия, после чего высушивают в сушильном шкафу при 105 - 110 °С. Сухой образец взвешивают. Если образец был покрыт смесью парафина с канифолью, то после катодного травления, которое проводят в этом случае при комнатной температуре электролита, его погружают в кипящую дистиллированную воду и щеткой счищают покрытие. После этого образец пассивируют, как описано выше, сушат и взвешивают.
А.9 При высоком содержании в отложениях меди, на что указывает омеднение поверхности образцов (образование медно-красного налета), операцию снятия отложений следует повторить с другим, неиспользованным образцом, предварительно поместив его (подготовленный и взвешенный) в раствор аммиака с массовой концентрацией 0,05 г/см3, содержащий 10 % персульфата аммония. После растворения меди образец промывают дистиллированной водой и помещают в раствор электролита для снятия отложений.
В тех случаях, когда другого образца нет, можно исправить результат, удалив налет металлической меди с обработанного образца. Для этого его помещают в аммиачный раствор персульфата аммония на 10 - 20 мин, после чего обмывают дистиллированной водой, ацетоном или спиртом, высушивают и взвешивают.
А.10 Загрязненность поверхности образца (q) определяют по формуле
![]()
где А - вес образца до обработки, г;
N - вес образца после обработки, г;
S - площадь очищенной поверхности, см2.
СОДЕРЖАНИЕ